

IDPicker 2.0: Improved protein assembly with high discrimination peptide identification filtering
- PMID: 19522537
- PMCID: PMC2810655
- DOI: 10.1021/pr900360j
IDPicker 2.0: Improved protein assembly with high discrimination peptide identification filtering
Abstract
Tandem mass spectrometry-based shotgun proteomics has become a widespread technology for analyzing complex protein mixtures. A number of database searching algorithms have been developed to assign peptide sequences to tandem mass spectra. Assembling the peptide identifications to proteins, however, is a challenging issue because many peptides are shared among multiple proteins. IDPicker is an open-source protein assembly tool that derives a minimum protein list from peptide identifications filtered to a specified False Discovery Rate. Here, we update IDPicker to increase confident peptide identifications by combining multiple scores produced by database search tools. By segregating peptide identifications for thresholding using both the precursor charge state and the number of tryptic termini, IDPicker retrieves more peptides for protein assembly. The new version is more robust against false positive proteins, especially in searches using multispecies databases, by requiring additional novel peptides in the parsimony process. IDPicker has been designed for incorporation in many identification workflows by the addition of a graphical user interface and the ability to read identifications from the pepXML format. These advances position IDPicker for high peptide discrimination and reliable protein assembly in large-scale proteomics studies. The source code and binaries for the latest version of IDPicker are available from http://fenchurch.mc.vanderbilt.edu/ .
Figures





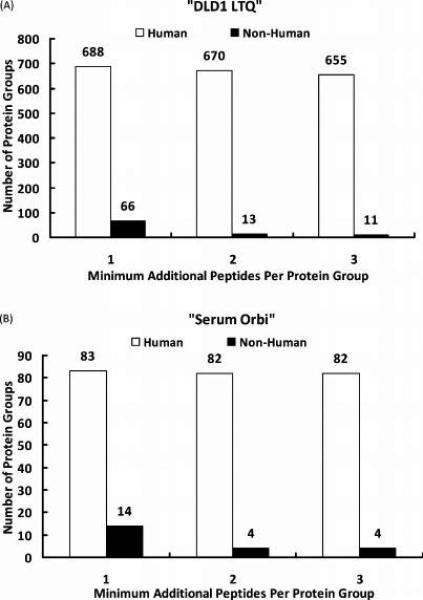
References
-
- Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422(6928):198–207. - PubMed
-
- Domon B, Aebersold R. Mass spectrometry and protein analysis. Science. 2006;312(5771):212–217. - PubMed
-
- Eng JKM,AL, Yates JR. An approach to correlate tandem mass-spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994;(5):976–989. - PubMed
-
- Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20(18):3551–3567. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
