

Prevalence of transcription promoters within archaeal operons and coding sequences
- PMID: 19536208
- PMCID: PMC2710873
- DOI: 10.1038/msb.2009.42
Prevalence of transcription promoters within archaeal operons and coding sequences
Abstract
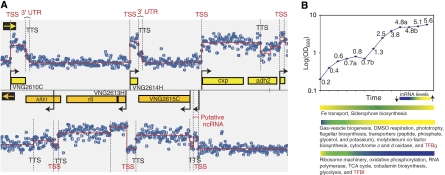
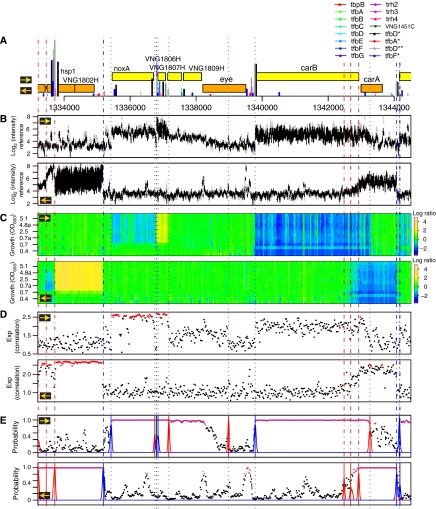
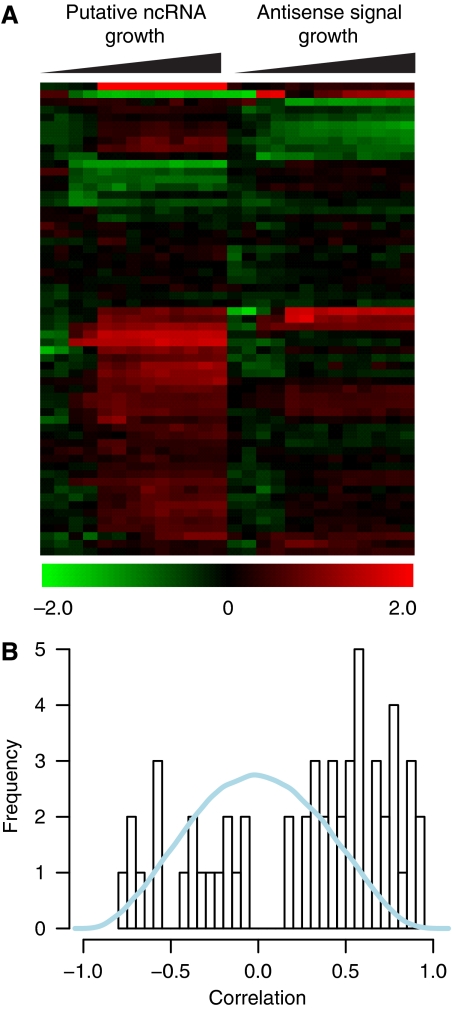
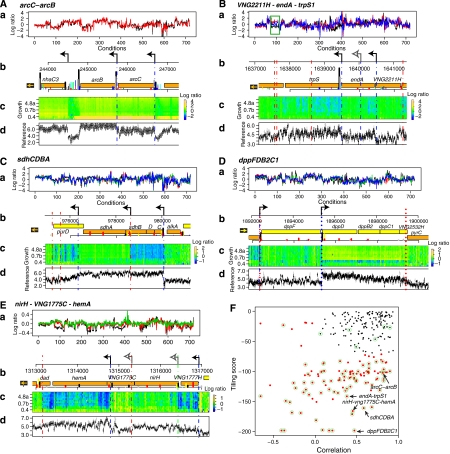
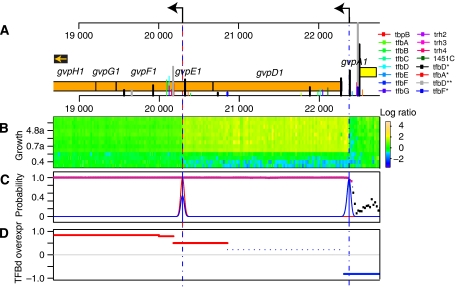
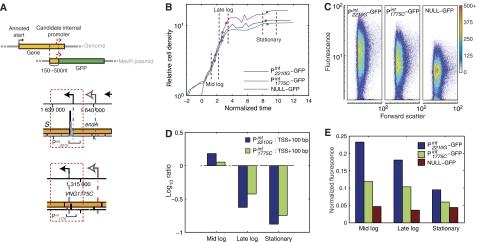
Despite the knowledge of complex prokaryotic-transcription mechanisms, generalized rules, such as the simplified organization of genes into operons with well-defined promoters and terminators, have had a significant role in systems analysis of regulatory logic in both bacteria and archaea. Here, we have investigated the prevalence of alternate regulatory mechanisms through genome-wide characterization of transcript structures of approximately 64% of all genes, including putative non-coding RNAs in Halobacterium salinarum NRC-1. Our integrative analysis of transcriptome dynamics and protein-DNA interaction data sets showed widespread environment-dependent modulation of operon architectures, transcription initiation and termination inside coding sequences, and extensive overlap in 3' ends of transcripts for many convergently transcribed genes. A significant fraction of these alternate transcriptional events correlate to binding locations of 11 transcription factors and regulators (TFs) inside operons and annotated genes-events usually considered spurious or non-functional. Using experimental validation, we illustrate the prevalence of overlapping genomic signals in archaeal transcription, casting doubt on the general perception of rigid boundaries between coding sequences and regulatory elements.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
