

The carboxyl terminus of Brca2 links the disassembly of Rad51 complexes to mitotic entry
- PMID: 19540122
- PMCID: PMC2719694
- DOI: 10.1016/j.cub.2009.05.057
The carboxyl terminus of Brca2 links the disassembly of Rad51 complexes to mitotic entry
Abstract
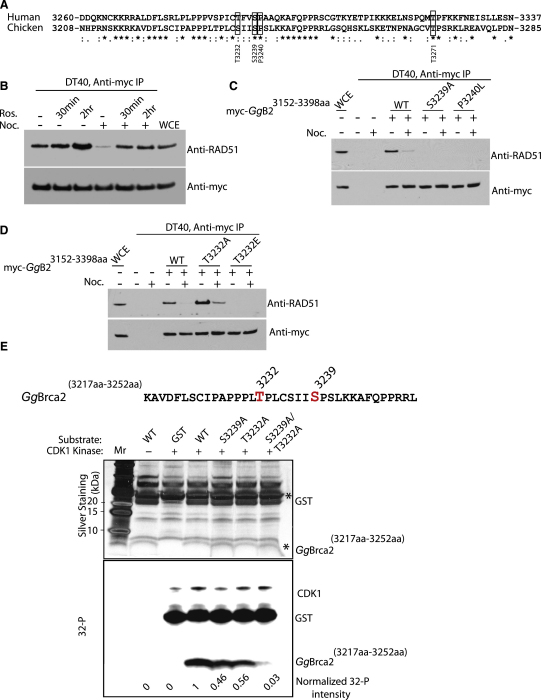
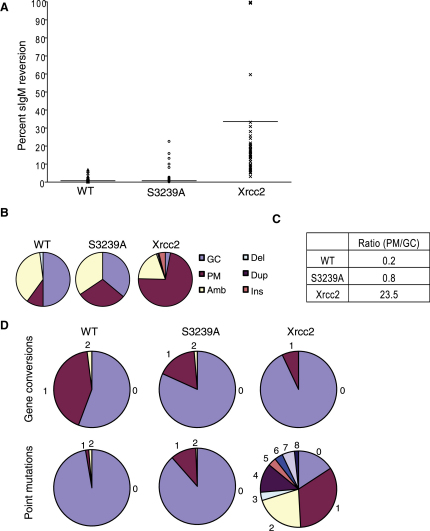
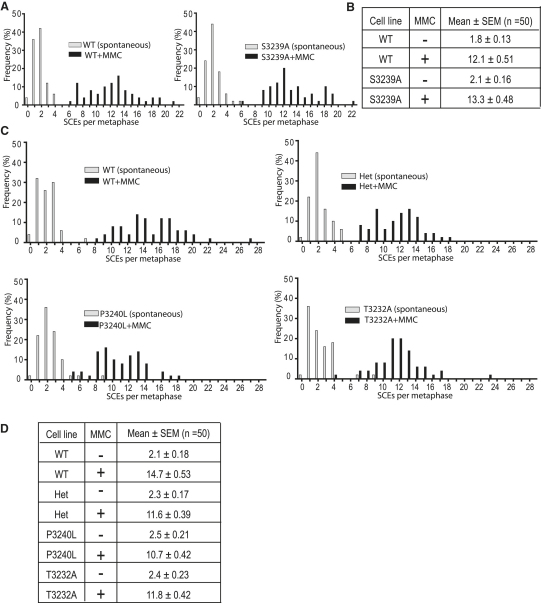
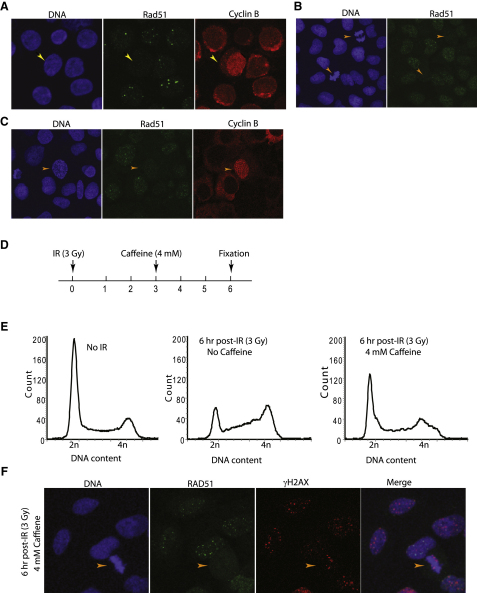
Background: The Rad51 recombinase assembles on DNA to execute homologous DNA recombination (HR). This process is essential to repair replication-associated genomic lesions before cells enter mitosis, but how it is started and stopped during the cell cycle remains poorly understood. Rad51 assembly is regulated by the breast cancer suppressor Brca2, via its evolutionarily conserved BRC repeats, and a distinct carboxy (C)-terminal motif whose biological function is uncertain. Using "hit-and-run" gene targeting to insert single-codon substitutions into the avian Brca2 locus, we report here a previously unrecognized role for the C-terminal motif.
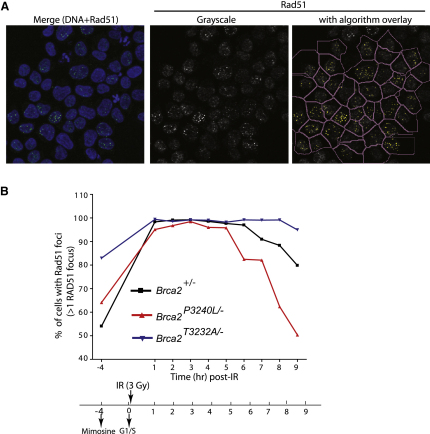
Results: We show that the avian C-terminal motif is functionally cognate with its human counterpart and identify point mutations that either abolish or enhance Rad51 binding. When these mutations are introduced into Brca2, we find that they affect neither the assembly of Rad51 into nuclear foci on damaged DNA nor DNA repair by HR. Instead, foci disassemble more rapidly in a point mutant that fails to bind Rad51, associated with faster mitotic entry. Conversely, the slower disassembly of foci in a point mutant that constitutively binds Rad51 correlates with delayed mitosis. Indeed, Rad51 foci do not persist in mitotic cells even after G2 checkpoint suppression, suggesting that their disassembly is a prerequisite for chromosome segregation.
Conclusions: We conclude that Rad51 binding by the C-terminal Brca2 motif is dispensable for the execution of HR but instead links the disassembly of Rad51 complexes to mitotic entry. This mechanism may ensure that HR terminates before chromosome segregation. Our findings assign a biological function for the C-terminal Brca2 motif in a mechanism that coordinates DNA repair with the cell cycle.
Figures

References
-
- Branzei D., Foiani M. Interplay of replication checkpoints and repair proteins at stalled replication forks. DNA Repair (Amst.) 2007;6:994–1003. - PubMed
-
- Cox M.M. The nonmutagenic repair of broken replication forks via recombination. Mutat. Res. 2002;510:107–120. - PubMed
-
- Haber J.E. DNA recombination: The replication connection. Trends Biochem. Sci. 1999;24:271–275. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
