

Interferon-beta treatment increases human papillomavirus early gene transcription and viral plasmid genome replication by activating interferon regulatory factor (IRF)-1
- PMID: 19541854
- PMCID: PMC7110192
- DOI: 10.1093/carcin/bgp150
Interferon-beta treatment increases human papillomavirus early gene transcription and viral plasmid genome replication by activating interferon regulatory factor (IRF)-1
Abstract
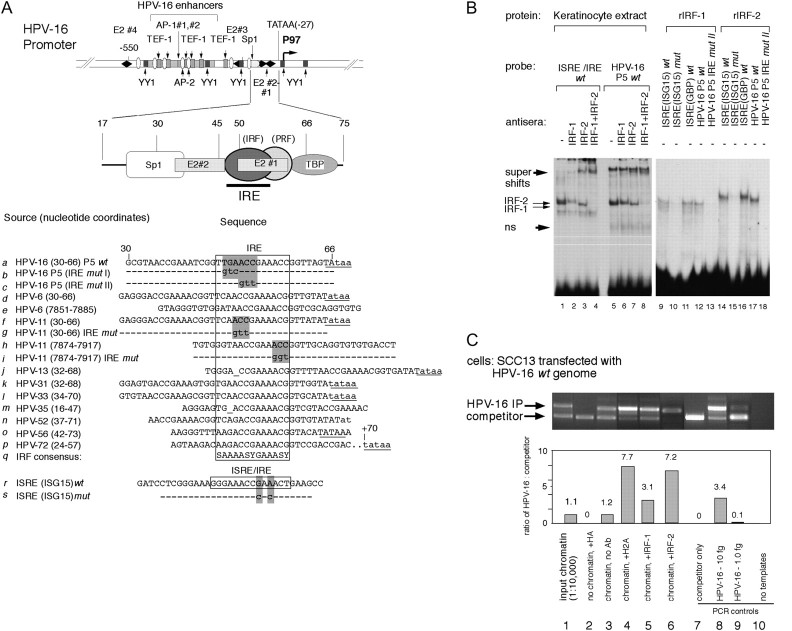
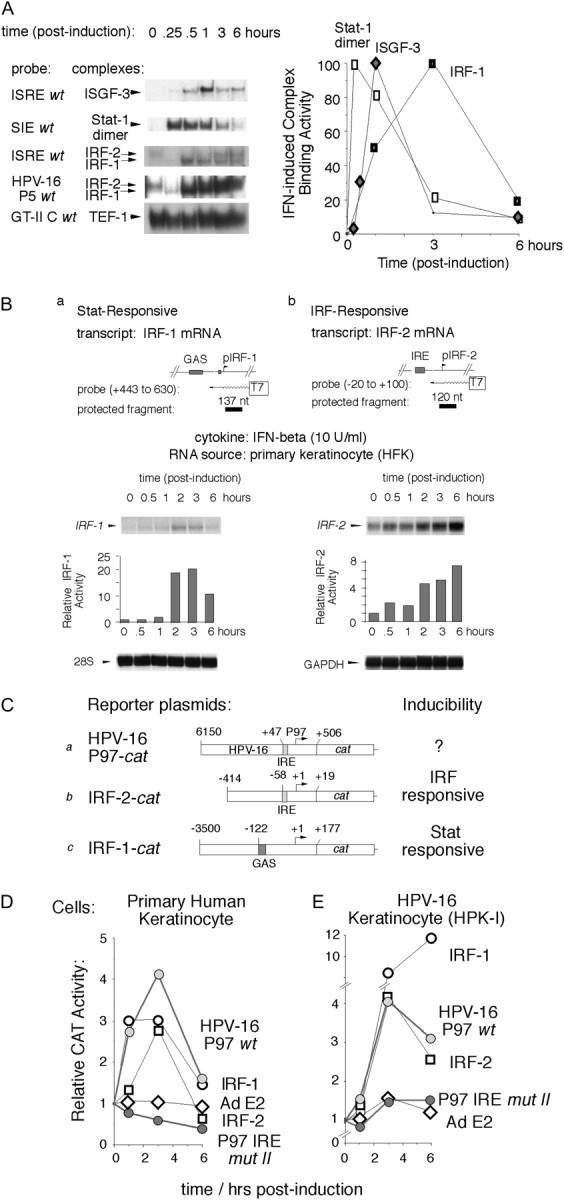
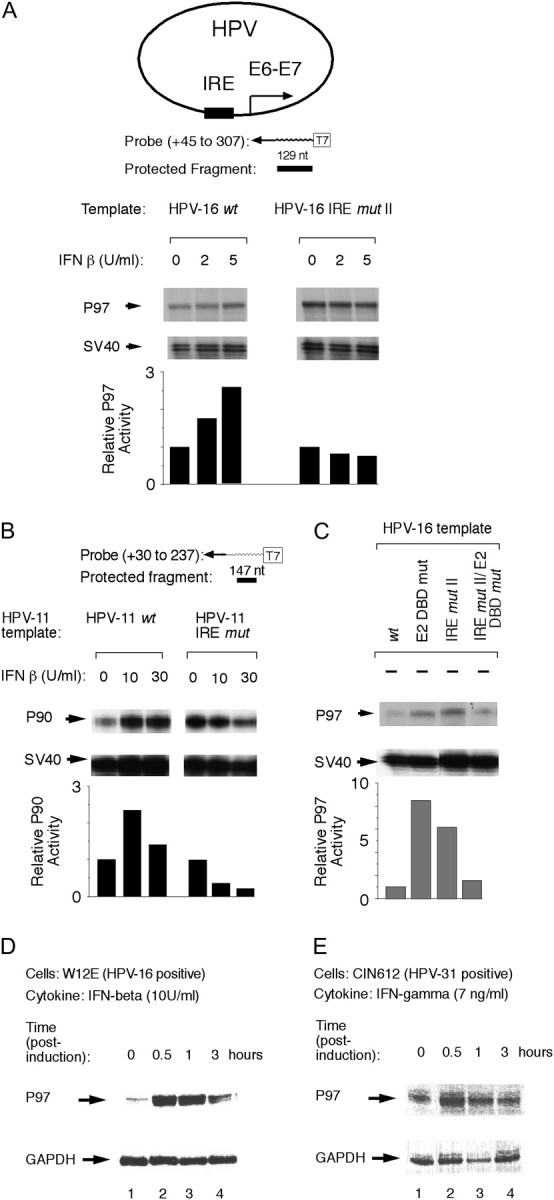
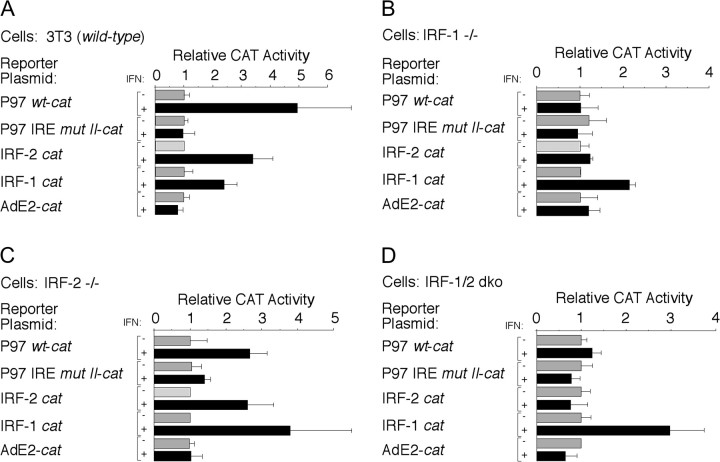
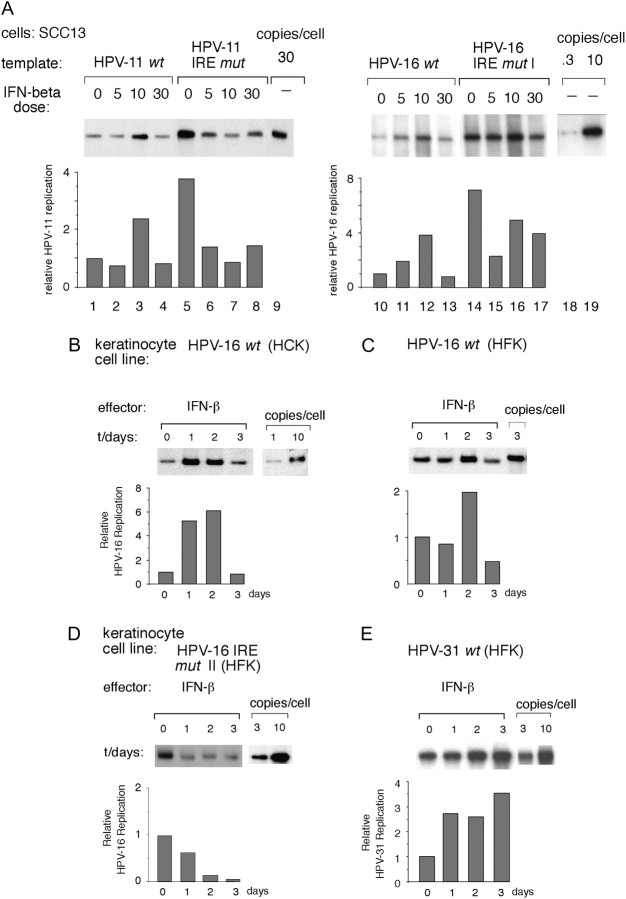
Interferons (IFNs) have been used to treat mucosal lesions caused by human papillomavirus (HPV) infection, such as intraepithelial precursor lesions to cancer of the uterine cervix, genital warts or recurrent respiratory papillomatosis, to potentially reduce or eliminate replicating HPV plasmid genomes. Mucosal HPVs have evolved mechanisms that impede IFN-beta synthesis and downregulate genes induced by IFN. Here we show that these HPV types directly subvert a cellular transcriptional response to IFN-beta as a potential boost in infection. Treatment with low levels of human IFN-beta induced initial amplification of HPV-16 and HPV-11 plasmid genomes and increased HPV-16 or HPV-31 DNA copy numbers up to 6-fold in HPV-immortalized keratinocytes. IFN treatment also increased early gene transcription from the major early gene promoters in HPV-16, HPV-31 and HPV-11. Furthermore, mutagenesis of the viral genomes and ectopic interferon regulatory factor (IRF) expression in transfection experiments using IRF-1(-/-), IRF-2(-/-) and dual knockout cell lines determined that these responses are due to the activation of IRF-1 interaction with a conserved interferon response element demonstrated in several mucosal HPV early gene promoters. Our results provide a molecular explanation for the varying clinical outcomes of IFN therapy of papillomatoses and define an assay for the modulation of the HPV gene program by IFNs as well as other cytokines and signaling molecules in infection and therapy.
Figures
Similar articles
-
Interferon regulatory factor (IRF)-2 activates the HPV-16 E6-E7 promoter in keratinocytes.Virology. 2010 Apr 10;399(2):270-9. doi: 10.1016/j.virol.2009.12.025. Epub 2010 Feb 2. Virology. 2010. PMID: 20129639
-
Interferon treatment of human keratinocytes harboring extrachromosomal, persistent HPV-16 plasmid genomes induces de novo viral integration.Carcinogenesis. 2015 Jan;36(1):151-9. doi: 10.1093/carcin/bgu236. Epub 2014 Nov 21. Carcinogenesis. 2015. PMID: 25416558
-
Human papillomavirus type 16 E5 protein induces expression of beta interferon through interferon regulatory factor 1 in human keratinocytes.J Virol. 2011 May;85(10):5070-80. doi: 10.1128/JVI.02114-10. Epub 2011 Mar 9. J Virol. 2011. PMID: 21389130 Free PMC article.
-
Role of Interferon Regulatory Factor 1 in acute and chronic virus infections.Virology. 2025 Feb;603:110386. doi: 10.1016/j.virol.2024.110386. Epub 2024 Dec 30. Virology. 2025. PMID: 39754861 Review.
-
Key role of interferon regulatory factor 1 (IRF-1) in regulating liver disease: progress and outlook.J Zhejiang Univ Sci B. 2024 Jun 15;25(6):451-470. doi: 10.1631/jzus.B2300159. J Zhejiang Univ Sci B. 2024. PMID: 38910492 Free PMC article. Review.
Cited by
-
The use of a human papillomavirus 18 promoter for tissue-specific expression in cervical carcinoma cells.Cell Mol Biol Lett. 2011 Sep;16(3):477-92. doi: 10.2478/s11658-011-0018-8. Epub 2011 Jul 18. Cell Mol Biol Lett. 2011. PMID: 21786035 Free PMC article.
-
Human papillomavirus (HPV) type 18 induces extended growth in primary human cervical, tonsillar, or foreskin keratinocytes more effectively than other high-risk mucosal HPVs.J Virol. 2009 Nov;83(22):11784-94. doi: 10.1128/JVI.01370-09. Epub 2009 Sep 9. J Virol. 2009. PMID: 19740985 Free PMC article.
-
Anticancer effects of the engineered stem cells transduced with therapeutic genes via a selective tumor tropism caused by vascular endothelial growth factor toward HeLa cervical cancer cells.Mol Cells. 2013 Oct;36(4):347-54. doi: 10.1007/s10059-013-0153-3. Epub 2013 Sep 2. Mol Cells. 2013. PMID: 24008363 Free PMC article.
-
The Interaction Between Human Papillomaviruses and the Stromal Microenvironment.Prog Mol Biol Transl Sci. 2016;144:169-238. doi: 10.1016/bs.pmbts.2016.09.003. Epub 2016 Oct 11. Prog Mol Biol Transl Sci. 2016. PMID: 27865458 Free PMC article. Review.
-
Study of serum level of kisspeptin and interferon-beta in genital wart patients.Indian J Sex Transm Dis AIDS. 2023 Jan-Jun;44(1):30-34. doi: 10.4103/ijstd.ijstd_93_22. Epub 2023 Jun 6. Indian J Sex Transm Dis AIDS. 2023. PMID: 37457538 Free PMC article.
References
-
- Psyrri A, et al. Human papillomavirus in cervical and head-and-neck cancer. Nat. Clin. Pract. Oncol. 2008;5:24–31. - PubMed
-
- Munoz-Fontela C, et al. Control of virus infection by tumour suppressors. Carcinogenesis. 2007;28:1140–1144. - PubMed
-
- Garcia-Sastre A, et al. Type 1 interferons and the virus-host relationship: a lesson in detente. Science. 2006;312:879–882. - PubMed
-
- Kimberlin DW. Current status of antiviral therapy for juvenile-onset recurrent respiratory papillomatosis. Antiviral Res. 2004;63:141–151. - PubMed
-
- Szeps M, et al. Human papillomavirus, viral load and proliferation rate in recurrent respiratory papillomatosis in response to alpha interferon treatment. J. Gen. Virol. 2005;86:1695–1702. - PubMed
