

Next-generation tag sequencing for cancer gene expression profiling
- PMID: 19541910
- PMCID: PMC2765282
- DOI: 10.1101/gr.094482.109
Next-generation tag sequencing for cancer gene expression profiling
Abstract
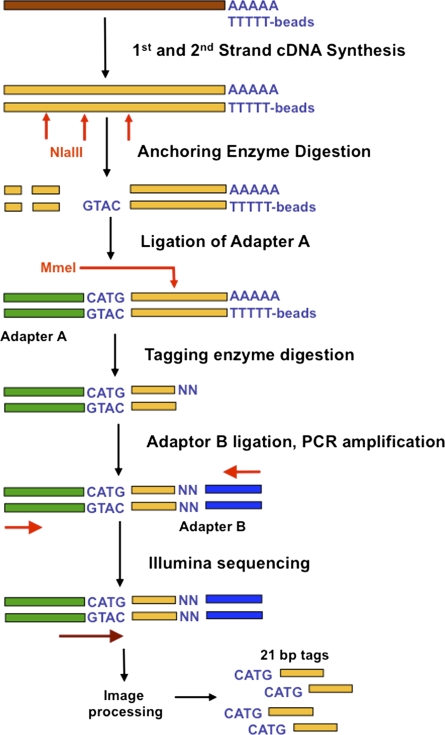
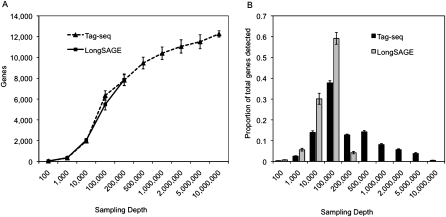
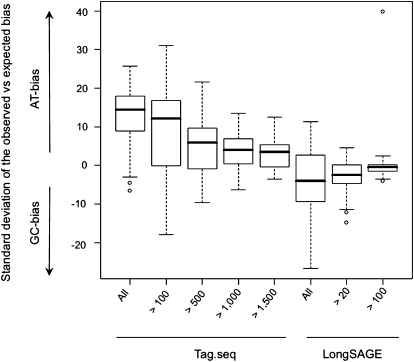
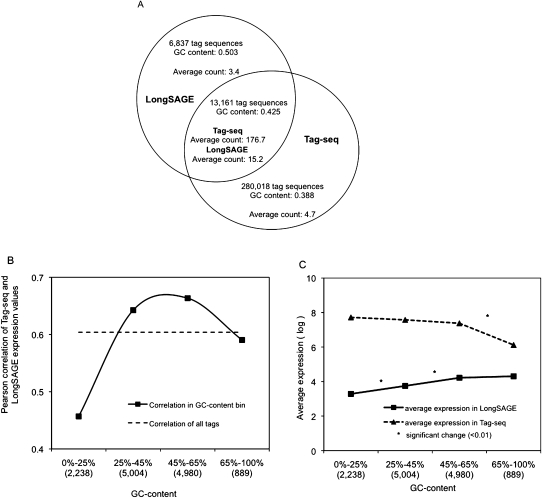
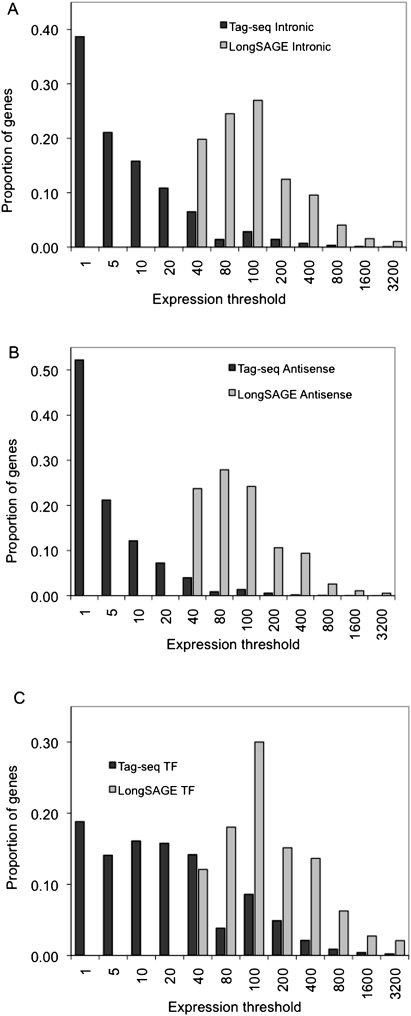
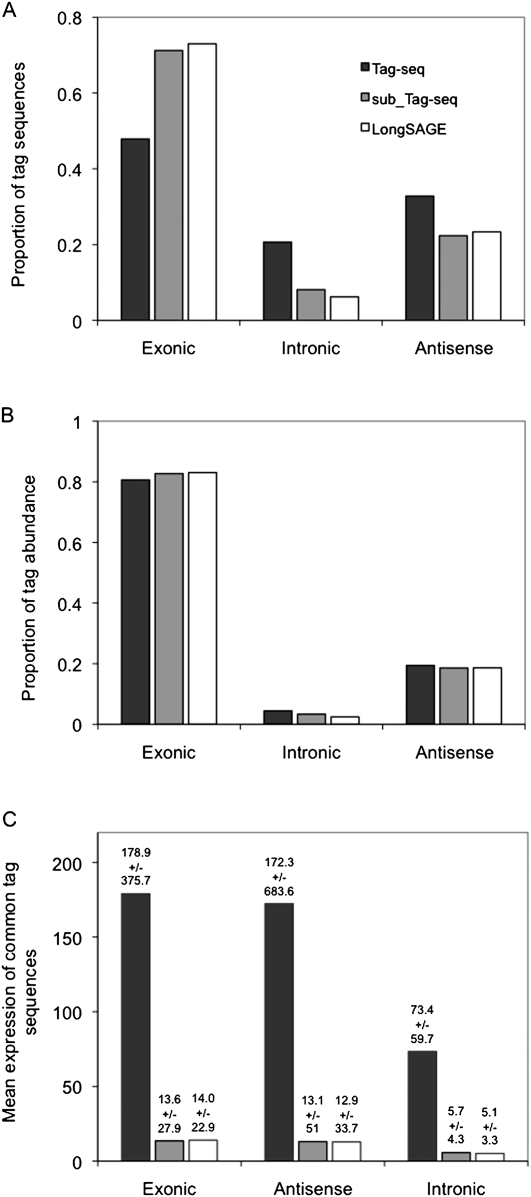
We describe a new method, Tag-seq, which employs ultra high-throughput sequencing of 21 base pair cDNA tags for sensitive and cost-effective gene expression profiling. We compared Tag-seq data to LongSAGE data and observed improved representation of several classes of rare transcripts, including transcription factors, antisense transcripts, and intronic sequences, the latter possibly representing novel exons or genes. We observed increases in the diversity, abundance, and dynamic range of such rare transcripts and took advantage of the greater dynamic range of expression to identify, in cancers and normal libraries, altered expression ratios of alternative transcript isoforms. The strand-specific information of Tag-seq reads further allowed us to detect altered expression ratios of sense and antisense (S-AS) transcripts between cancer and normal libraries. S-AS transcripts were enriched in known cancer genes, while transcript isoforms were enriched in miRNA targeting sites. We found that transcript abundance had a stronger GC-bias in LongSAGE than Tag-seq, such that AT-rich tags were less abundant than GC-rich tags in LongSAGE. Tag-seq also performed better in gene discovery, identifying >98% of genes detected by LongSAGE and profiling a distinct subset of the transcriptome characterized by AT-rich genes, which was expressed at levels below those detectable by LongSAGE. Overall, Tag-seq is sensitive to rare transcripts, has less sequence composition bias relative to LongSAGE, and allows differential expression analysis for a greater range of transcripts, including transcripts encoding important regulatory molecules.
Figures
References
-
- Adams MD, Kelley JM, Gocayne JD, Dubnick M, Polymeropoulos MH, Xiao H, Merril CR, Wu A, Olde B, Moreno RF, et al. Complementary DNA sequencing: Expressed sequence tags and human genome project. Science. 1991;252:1651–1656. - PubMed
-
- Bentley DR. Whole-genome re-sequencing. Curr Opin Genet Dev. 2006;16:545–552. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous