

PGC-1{alpha} and PGC-1{beta} regulate mitochondrial density in neurons
- PMID: 19542216
- PMCID: PMC2755862
- DOI: 10.1074/jbc.M109.018911
PGC-1{alpha} and PGC-1{beta} regulate mitochondrial density in neurons
Abstract
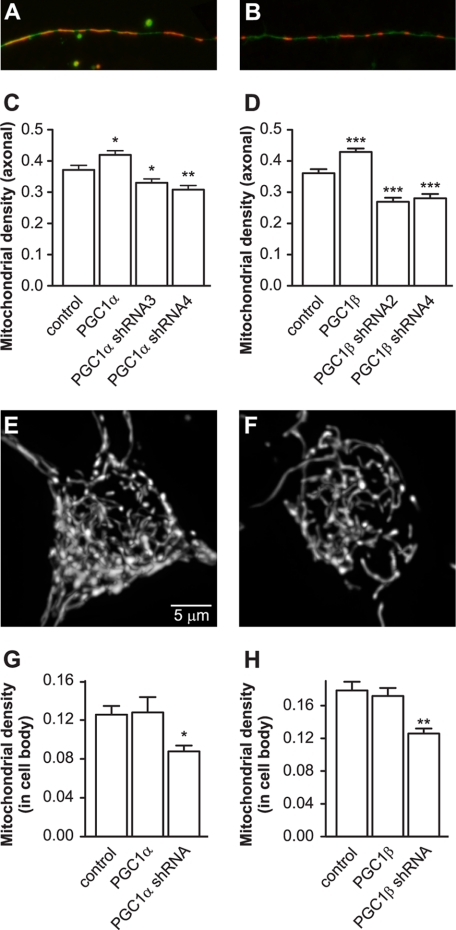
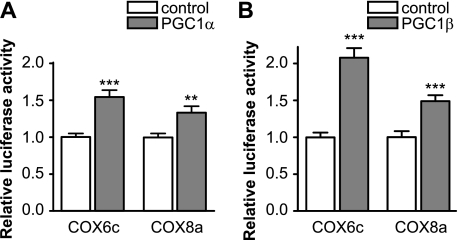
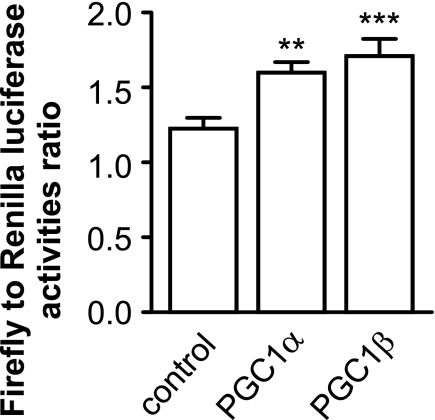
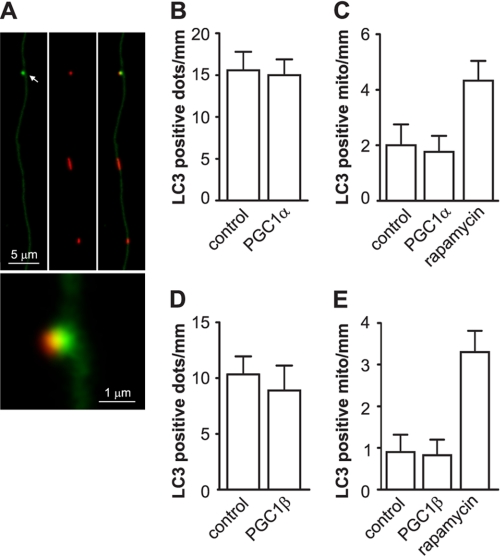
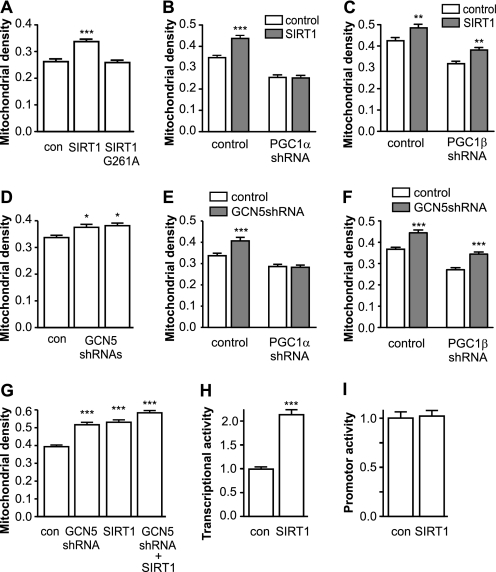
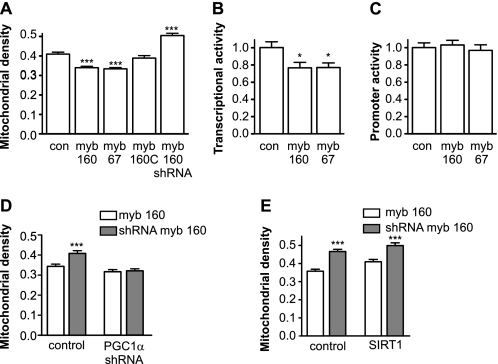
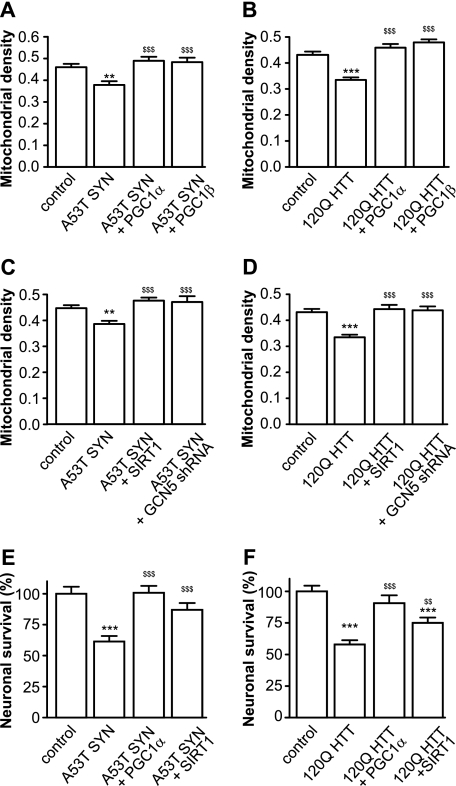
Recent studies indicate that regulation of cellular oxidative capacity through enhancing mitochondrial biogenesis may be beneficial for neuronal recovery and survival in human neurodegenerative disorders. The peroxisome proliferator-activated receptor gamma coactivator-1alpha (PGC-1alpha) has been shown to be a master regulator of mitochondrial biogenesis and cellular energy metabolism in muscle and liver. The aim of our study was to establish whether PGC-1alpha and PGC-1beta control mitochondrial density also in neurons and if these coactivators could be up-regulated by deacetylation. The results demonstrate that PGC-1alpha and PGC-1beta control mitochondrial capacity in an additive and independent manner. This effect was observed in all studied subtypes of neurons, in cortical, midbrain, and cerebellar granule neurons. We also observed that endogenous neuronal PGC-1alpha but not PGC-1beta could be activated through its repressor domain by suppressing it. Results demonstrate also that overexpression of SIRT1 deacetylase or suppression of GCN5 acetyltransferase activates transcriptional activity of PGC-1alpha in neurons and increases mitochondrial density. These effects were mediated exclusively via PGC-1alpha, since overexpression of SIRT1 or suppression of GCN5 was ineffective where PGC-1alpha was suppressed by short hairpin RNA. Moreover, the results demonstrate that overexpression of PGC-1beta or PGC-1alpha or activation of the latter by SIRT1 protected neurons from mutant alpha-synuclein- or mutant huntingtin-induced mitochondrial loss. These evidences demonstrate that activation or overexpression of the PGC-1 family of coactivators could be used to compensate for neuronal mitochondrial loss and suggest that therapeutic agents activating PGC-1 would be valuable for treating neurodegenerative diseases in which mitochondrial dysfunction and oxidative damage play an important pathogenic role.
Figures
Similar articles
-
Sirtuin 1 (SIRT1) deacetylase activity is not required for mitochondrial biogenesis or peroxisome proliferator-activated receptor-gamma coactivator-1alpha (PGC-1alpha) deacetylation following endurance exercise.J Biol Chem. 2011 Sep 2;286(35):30561-30570. doi: 10.1074/jbc.M111.261685. Epub 2011 Jul 11. J Biol Chem. 2011. PMID: 21757760 Free PMC article.
-
The deacetylase enzyme SIRT1 is not associated with oxidative capacity in rat heart and skeletal muscle and its overexpression reduces mitochondrial biogenesis.J Physiol. 2009 Apr 15;587(Pt 8):1817-28. doi: 10.1113/jphysiol.2008.168096. Epub 2009 Feb 23. J Physiol. 2009. PMID: 19237425 Free PMC article.
-
Deacetylation of PGC-1α by SIRT1: importance for skeletal muscle function and exercise-induced mitochondrial biogenesis.Appl Physiol Nutr Metab. 2011 Oct;36(5):589-97. doi: 10.1139/h11-070. Epub 2011 Sep 2. Appl Physiol Nutr Metab. 2011. PMID: 21888529 Review.
-
The diabetes medication canagliflozin promotes mitochondrial remodelling of adipocyte via the AMPK-Sirt1-Pgc-1α signalling pathway.Adipocyte. 2020 Dec;9(1):484-494. doi: 10.1080/21623945.2020.1807850. Adipocyte. 2020. PMID: 32835596 Free PMC article.
-
PGC-1α, mitochondrial dysfunction, and Huntington's disease.Free Radic Biol Med. 2013 Sep;62:37-46. doi: 10.1016/j.freeradbiomed.2013.04.016. Epub 2013 Apr 19. Free Radic Biol Med. 2013. PMID: 23602910 Free PMC article. Review.
Cited by
-
Genetic association of sirtuin genes and Parkinson's disease.J Neurol. 2013 Sep;260(9):2237-41. doi: 10.1007/s00415-013-6970-7. Epub 2013 May 30. J Neurol. 2013. PMID: 23719790
-
PGC-1α activity in nigral dopamine neurons determines vulnerability to α-synuclein.Acta Neuropathol Commun. 2015 Apr 1;3:16. doi: 10.1186/s40478-015-0200-8. Acta Neuropathol Commun. 2015. PMID: 25853296 Free PMC article.
-
Mitochondria and Oxidative Stress as a Link between Alzheimer's Disease and Diabetes Mellitus.Int J Mol Sci. 2023 Sep 22;24(19):14450. doi: 10.3390/ijms241914450. Int J Mol Sci. 2023. PMID: 37833898 Free PMC article. Review.
-
HDAC inhibition induces autophagy and mitochondrial biogenesis to maintain mitochondrial homeostasis during cardiac ischemia/reperfusion injury.J Mol Cell Cardiol. 2019 May;130:36-48. doi: 10.1016/j.yjmcc.2019.03.008. Epub 2019 Mar 14. J Mol Cell Cardiol. 2019. PMID: 30880250 Free PMC article.
-
Roles of oxidative stress, apoptosis, PGC-1α and mitochondrial biogenesis in cerebral ischemia.Int J Mol Sci. 2011;12(10):7199-215. doi: 10.3390/ijms12107199. Epub 2011 Oct 21. Int J Mol Sci. 2011. PMID: 22072942 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
