

Mechanism and regulatory function of CpG signaling via scavenger receptor B1 in primary B cells
- PMID: 19542230
- PMCID: PMC2755695
- DOI: 10.1074/jbc.M109.018580
Mechanism and regulatory function of CpG signaling via scavenger receptor B1 in primary B cells
Abstract
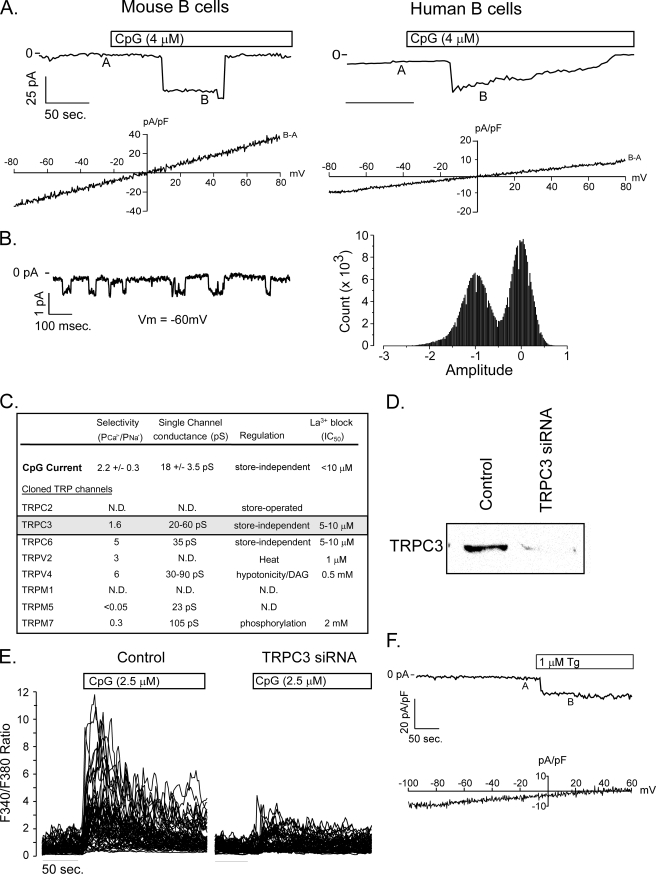
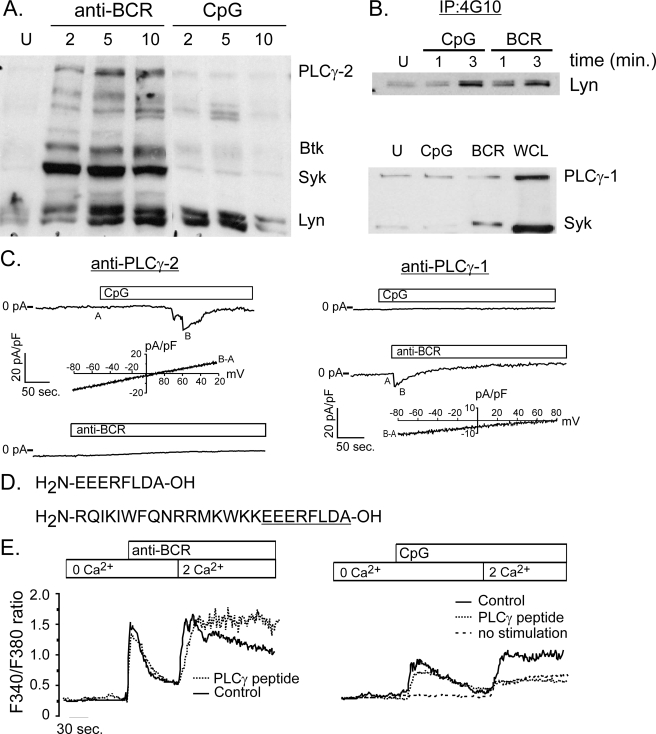
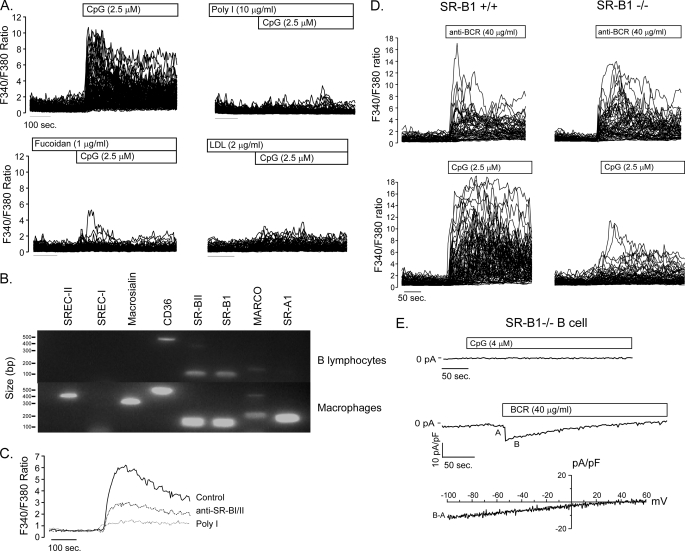
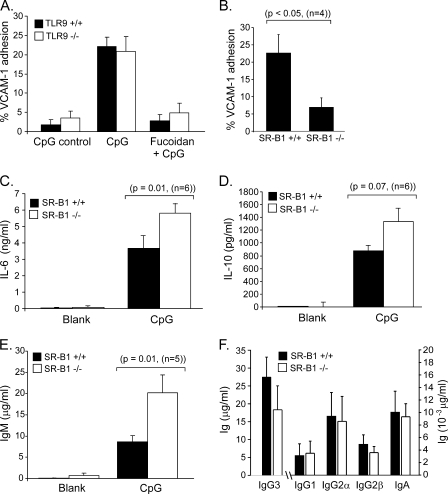
It is well established that CpG promotes pro-inflammatory cytokine and antibody production by B cells via the Toll-like receptor 9 (TLR9)-dependent pathway. However, scavenger receptors (SRs) are also capable of binding such pathogen-derived molecules, yet their contribution to CpG-induced signaling events has not yet been evaluated. Here we identified a novel TLR9-independent mechanism of CpG-induced signaling and immune function that is mediated by the scavenger B1 receptor (SR-B1). Specifically, we show that CpG/SR-B1 triggers calcium entry into primary B lymphocytes via phospholipase C gamma-1-mediated activation of TRPC3 channels and also B cell adhesion to vascular cell adhesion molecule-1. CpG-induced calcium signals and vascular cell adhesion molecule-1 adhesion are TLR9-independent and are mediated exclusively by SR-B1. Although pro-inflammatory cytokine and Ig production induced by CpG require TLR9 expression, we also found that SR-B1 negatively regulates TLR9-dependent production of interleukin-6, interleukin-10, and IgM. Thus, our results provide a novel perspective on the complexity of CpG signaling within B cells by demonstrating that SR-B1 is an alternative pathway for nucleic acid-induced signaling that provides feedback inhibition on specific TLR9-dependent responses of B cells. Consequently, these results have wide implications for understanding the mechanisms regulating immune tolerance to nucleic acids and pathogen-associated molecules.
Figures

Similar articles
-
B cell TLR1/2, TLR4, TLR7 and TLR9 interact in induction of class switch DNA recombination: modulation by BCR and CD40, and relevance to T-independent antibody responses.Autoimmunity. 2015 Feb;48(1):1-12. doi: 10.3109/08916934.2014.993027. Autoimmunity. 2015. PMID: 25536171 Free PMC article.
-
Toll-like receptor 9 signaling by CpG-B oligodeoxynucleotides induces an apoptotic pathway in human chronic lymphocytic leukemia B cells.Blood. 2010 Jun 17;115(24):5041-52. doi: 10.1182/blood-2009-03-213363. Epub 2010 Mar 25. Blood. 2010. PMID: 20339095 Free PMC article.
-
Bruton's tyrosine kinase separately regulates NFkappaB p65RelA activation and cytokine interleukin (IL)-10/IL-12 production in TLR9-stimulated B Cells.J Biol Chem. 2008 Apr 25;283(17):11189-98. doi: 10.1074/jbc.M708516200. Epub 2008 Feb 13. J Biol Chem. 2008. PMID: 18276597
-
Exploiting scavenger receptors in cancer immunotherapy: Lessons from CD5 and SR-B1.Eur J Immunol. 2017 Jul;47(7):1108-1118. doi: 10.1002/eji.201646903. Epub 2017 Jun 12. Eur J Immunol. 2017. PMID: 28504304 Review.
-
Role of Scavenger Receptor B1 (SR-B1) in Improving Food Benefits for Human Health.Annu Rev Food Sci Technol. 2025 Apr;16(1):403-432. doi: 10.1146/annurev-food-111523-121935. Epub 2025 Feb 3. Annu Rev Food Sci Technol. 2025. PMID: 39899837 Review.
Cited by
-
Regulation of intestinal immunity by dietary fatty acids.Mucosal Immunol. 2022 May;15(5):846-856. doi: 10.1038/s41385-022-00547-2. Epub 2022 Jul 12. Mucosal Immunol. 2022. PMID: 35821290 Review.
-
Intracellular processing of immunostimulatory CpG-siRNA: Toll-like receptor 9 facilitates siRNA dicing and endosomal escape.J Control Release. 2013 Sep 28;170(3):307-15. doi: 10.1016/j.jconrel.2013.06.007. Epub 2013 Jun 15. J Control Release. 2013. PMID: 23777886 Free PMC article.
-
Syk is indispensable for CpG-induced activation and differentiation of human B cells.Cell Mol Life Sci. 2015 Jun;72(11):2223-36. doi: 10.1007/s00018-014-1806-x. Epub 2014 Dec 28. Cell Mol Life Sci. 2015. PMID: 25543269 Free PMC article.
-
B-lymphocyte calcium influx.Immunol Rev. 2009 Sep;231(1):265-77. doi: 10.1111/j.1600-065X.2009.00822.x. Immunol Rev. 2009. PMID: 19754903 Free PMC article. Review.
-
Influence of physicochemical properties of silver nanoparticles on mast cell activation and degranulation.Toxicol In Vitro. 2015 Feb;29(1):195-203. doi: 10.1016/j.tiv.2014.10.008. Toxicol In Vitro. 2015. PMID: 25458489 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
