

Early treatment with alglucosidase alpha prolongs long-term survival of infants with Pompe disease
- PMID: 19542901
- PMCID: PMC3129995
- DOI: 10.1203/PDR.0b013e3181b24e94
Early treatment with alglucosidase alpha prolongs long-term survival of infants with Pompe disease
Abstract
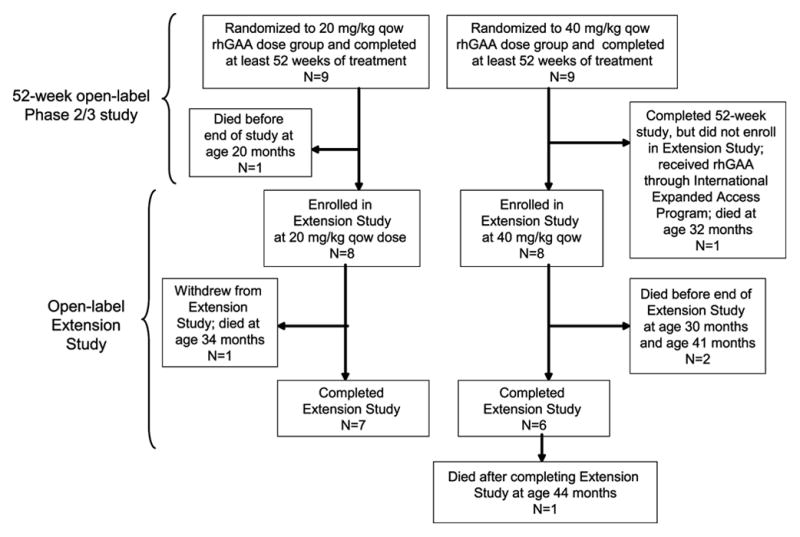
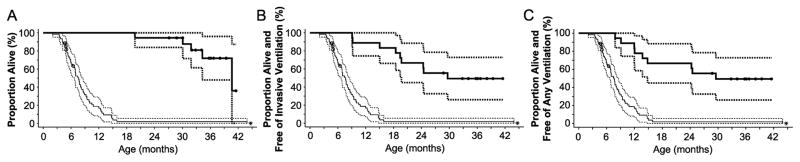
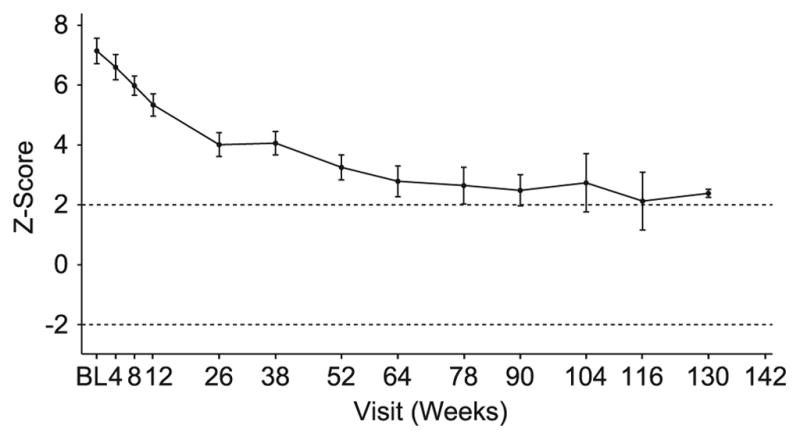
In a previous 52-wk trial, treatment with alglucosidase alpha markedly improved cardiomyopathy, ventilatory function, and overall survival among 18 children <7 mo old with infantile-onset Pompe disease. Sixteen of the 18 patients enrolled in an extension study, where they continued to receive alglucosidase alpha at either 20 mg/kg biweekly (n = 8) or 40 mg/kg biweekly (n = 8), for up to a total of 3 y. These children continued to exhibit the benefits of alglucosidase alpha at the age of 36 mo. Cox regression analyses showed that over the entire study period, alglucosidase alpha treatment reduced the risk of death by 95%, reduced the risk of invasive ventilation or death by 91%, and reduced the risk of any type of ventilation or death by 87%, compared with an untreated historical control group. Cardiomyopathy continued to improve and 11 patients learned and sustained substantial motor skills. No significant differences in either safety or efficacy parameters were observed between the 20 and 40 mg/kg biweekly doses. Overall, long-term alglucosidase alpha treatment markedly extended survival as well as ventilation-free survival and improved cardiomyopathy.
Figures
References
-
- Hirschhorn R, Reuser AJ. Glycogen storage disease type II: acid α-glucosidase (acid maltase) deficiency. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. 8. McGraw Hill; New York: 2001. pp. 3389–3420.
-
- Kishnani PS, Howell RR. Pompe disease in infants and children. J Pediatr. 2004;144:S35–S43. - PubMed
-
- Chen YT, Amalfitano A. Towards a molecular therapy for glycogen storage disease type II (Pompe disease) Mol Med Today. 2000;6:245–251. - PubMed
-
- van den Hout HM, Hop W, van Diggelen OP, Smeitink JA, Smit GP, Poll-The BT, Bakker HD, Loonen MC, de Klerk JB, Reuser AJ, van der Ploeg AT. The natural course of infantile Pompe’s disease: 20 original cases compared with 133 cases from the literature. Pediatrics. 2003;112:332–340. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
