

TGF-beta activates Akt kinase through a microRNA-dependent amplifying circuit targeting PTEN
- PMID: 19543271
- PMCID: PMC2744130
- DOI: 10.1038/ncb1897
TGF-beta activates Akt kinase through a microRNA-dependent amplifying circuit targeting PTEN
Abstract
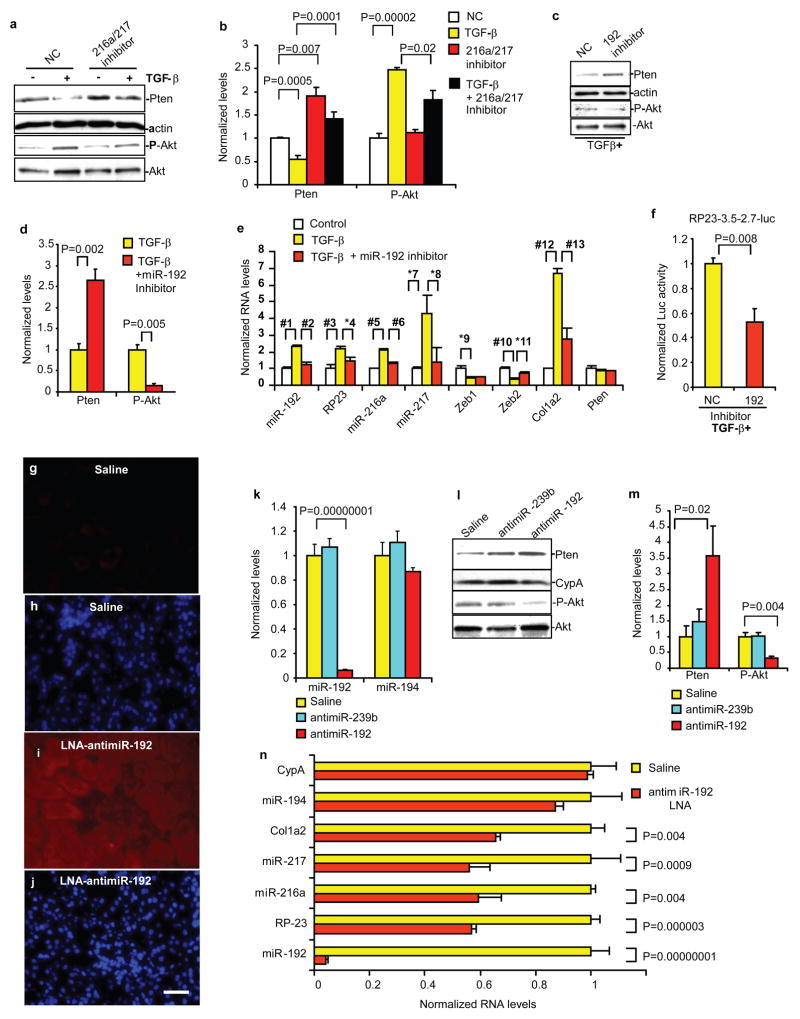
Akt kinase is activated by transforming growth factor-beta1 (TGF-beta) in diabetic kidneys, and has important roles in fibrosis, hypertrophy and cell survival in glomerular mesangial cells. However, the mechanisms of Akt activation by TGF-beta are not fully understood. Here we show that TGF-beta activates Akt in glomerular mesangial cells by inducing the microRNAs (miRNAs) miR-216a and miR-217, both of which target PTEN (phosphatase and tensin homologue), an inhibitor of Akt activation. These miRNAs are located within the second intron of a non-coding RNA (RP23-298H6.1-001). The RP23 promoter was activated by TGF-beta and miR-192 through E-box-regulated mechanisms, as shown previously. Akt activation by these miRs led to glomerular mesangial cell survival and hypertrophy, which were similar to the effects of activation by TGF-beta. These studies reveal a mechanism of Akt activation through PTEN downregulation by two miRs, which are regulated by upstream miR-192 and TGF-beta. Due to the diversity of PTEN function, this miR-amplifying circuit may have key roles, not only in kidney disorders, but also in other diseases.
Figures




References
-
- Sharma K, Ziyadeh FN. Hyperglycemia and diabetic kidney disease. The case for transforming growth factor-beta as a key mediator. Diabetes. 1995;44:1139–1146. - PubMed
-
- Kato M, et al. Role of the Akt/FoxO3a pathway in TGF-beta1-mediated mesangial cell dysfunction: a novel mechanism related to diabetic kidney disease. J Am Soc Nephrol. 2006;17:3325–3335. - PubMed
-
- Mahimainathan L, Das F, Venkatesan B, Choudhury GG. Mesangial cell hypertrophy by high glucose is mediated by downregulation of the tumor suppressor PTEN. Diabetes. 2006;55:2115–2125. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
