

Selective processing and metabolism of disease-causing mutant prion proteins
- PMID: 19543376
- PMCID: PMC2691595
- DOI: 10.1371/journal.ppat.1000479
Selective processing and metabolism of disease-causing mutant prion proteins
Abstract
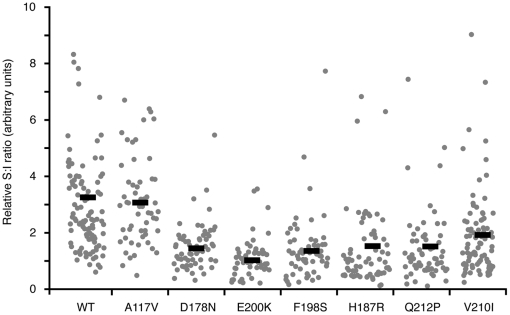
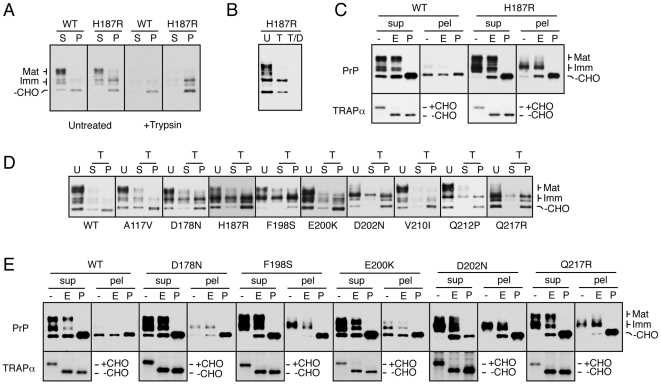
Prion diseases are fatal neurodegenerative disorders caused by aberrant metabolism of the cellular prion protein (PrP(C)). In genetic forms of these diseases, mutations in the globular C-terminal domain are hypothesized to favor the spontaneous generation of misfolded PrP conformers (including the transmissible PrP(Sc) form) that trigger downstream pathways leading to neuronal death. A mechanistic understanding of these diseases therefore requires knowledge of the quality control pathways that recognize and degrade aberrant PrPs. Here, we present comparative analyses of the biosynthesis, trafficking, and metabolism of a panel of genetic disease-causing prion protein mutants in the C-terminal domain. Using quantitative imaging and biochemistry, we identify a misfolded subpopulation of each mutant PrP characterized by relative detergent insolubility, inaccessibility to the cell surface, and incomplete glycan modifications. The misfolded populations of mutant PrPs were neither recognized by ER quality control pathways nor routed to ER-associated degradation despite demonstrable misfolding in the ER. Instead, mutant PrPs trafficked to the Golgi, from where the misfolded subpopulation was selectively trafficked for degradation in acidic compartments. Surprisingly, selective re-routing was dependent not only on a mutant globular domain, but on an additional lysine-based motif in the highly conserved unstructured N-terminus. These results define a specific trafficking and degradation pathway shared by many disease-causing PrP mutants. As the acidic lysosomal environment has been implicated in facilitating the conversion of PrP(C) to PrP(Sc), our identification of a mutant-selective trafficking pathway to this compartment may provide a cell biological basis for spontaneous generation of PrP(Sc) in familial prion disease.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures









References
-
- Gregersen N, Bross P, Vang S, Christensen JH. Protein misfolding and human disease. Annu Rev Genomics Hum Genet. 2006;7:103–124. - PubMed
-
- Arvan P, Zhao X, Ramos-Castaneda J, Chang A. Secretory pathway quality control operating in Golgi, plasmalemmal, and endosomal systems. Traffic. 2002;3:771–780. - PubMed
-
- Aguzzi A, Baumann F, Bremer J. The prion's elusive reason for being. Ann Rev Neurosci. 2008;31:439–477. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
