

Impairment of mitochondrial calcium handling in a mtSOD1 cell culture model of motoneuron disease
- PMID: 19545440
- PMCID: PMC2716351
- DOI: 10.1186/1471-2202-10-64
Impairment of mitochondrial calcium handling in a mtSOD1 cell culture model of motoneuron disease
Abstract
Background: Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disorder characterized by the selective loss of motor neurons (MN) in the brain stem and spinal cord. Intracellular disruptions of cytosolic and mitochondrial calcium have been associated with selective MN degeneration, but the underlying mechanisms are not well understood. The present evidence supports a hypothesis that mitochondria are a target of mutant SOD1-mediated toxicity in familial amyotrophic lateral sclerosis (fALS) and intracellular alterations of cytosolic and mitochondrial calcium might aggravate the course of this neurodegenerative disease. In this study, we used a fluorescence charged cool device (CCD) imaging system to separate and simultaneously monitor cytosolic and mitochondrial calcium concentrations in individual cells in an established cellular model of ALS.
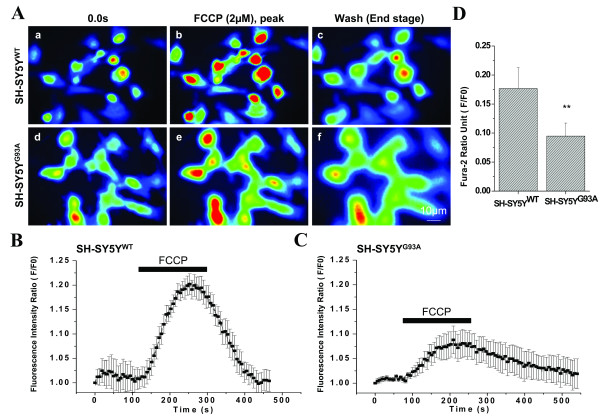
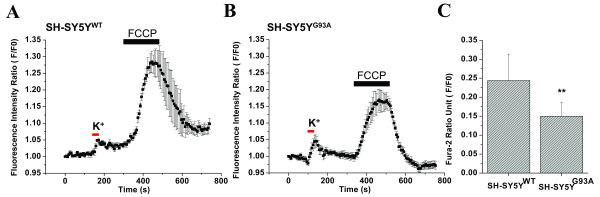
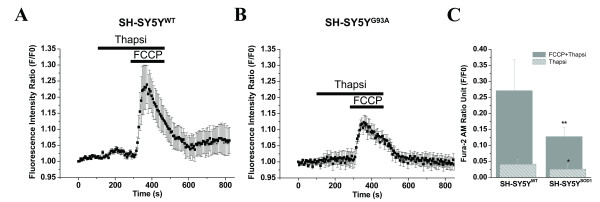
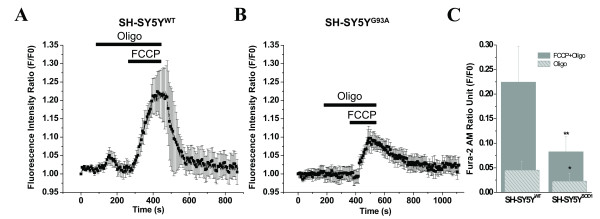
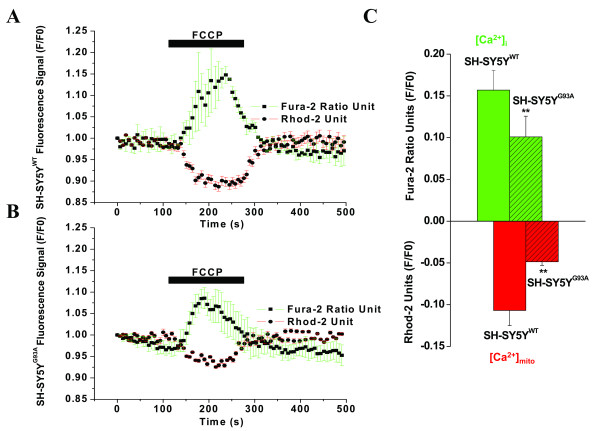
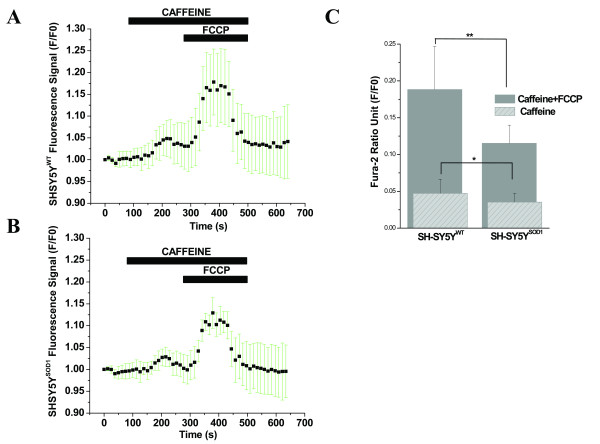
Results: To gain insights into the molecular mechanisms of SOD1(G93A) associated motor neuron disease, we simultaneously monitored cytosolic and mitochondrial calcium concentrations in individual cells. Voltage - dependent cytosolic Ca2+ elevations and mitochondria - controlled calcium release mechanisms were monitored after loading cells with fluorescent dyes fura-2 and rhod-2. Interestingly, comparable voltage-dependent cytosolic Ca2+ elevations in WT (SH-SY5Y(WT)) and G93A (SH-SY5Y(G93A)) expressing cells were observed. In contrast, mitochondrial intracellular Ca2+ release responses evoked by bath application of the mitochondrial toxin FCCP were significantly smaller in G93A expressing cells, suggesting impaired calcium stores. Pharmacological experiments further supported the concept that the presence of G93A severely disrupts mitochondrial Ca2+ regulation.
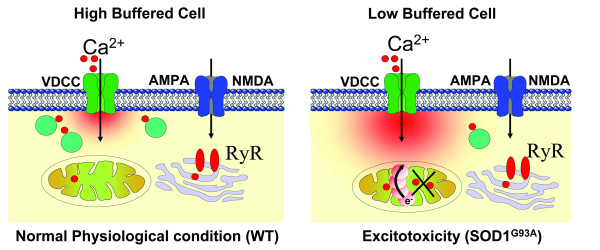
Conclusion: In this study, by fluorescence measurement of cytosolic calcium and using simultaneous [Ca2+]i and [Ca2+]mito measurements, we are able to separate and simultaneously monitor cytosolic and mitochondrial calcium concentrations in individual cells an established cellular model of ALS. The primary goals of this paper are (1) method development, and (2) screening for deficits in mutant cells on the single cell level. On the technological level, our method promises to serve as a valuable tool to identify mitochondrial and Ca2+-related defects during G93A-mediated MN degeneration. In addition, our experiments support a model where a specialized interplay between cytosolic calcium profiles and mitochondrial mechanisms contribute to the selective degeneration of neurons in ALS.
Figures
References
-
- Rosen DR, Bowling AC, Patterson D, Usdin TB, Sapp P, Mezey E, McKenna-Yasek D, O'Regan J, Rahmani Z, Ferrante RJ, et al. A frequent ala 4 to val superoxide dismutase-1 mutation is associated with a rapidly progressive familial amyotrophic lateral sclerosis. Hum Mol Genet. 1994;3:981–7. doi: 10.1093/hmg/3.6.981. - DOI - PubMed
-
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng H-X, Chen W, Zhai P, Sufit RL, Siddique T. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
