

Novel non-genomic signaling of thyroid hormone receptors in thyroid carcinogenesis
- PMID: 19549593
- PMCID: PMC2744088
- DOI: 10.1016/j.mce.2009.01.007
Novel non-genomic signaling of thyroid hormone receptors in thyroid carcinogenesis
Abstract
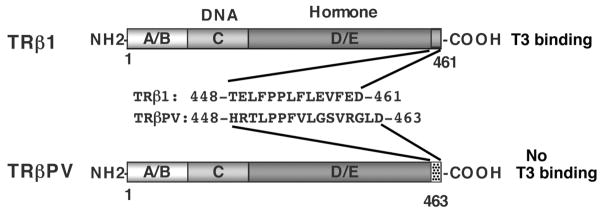
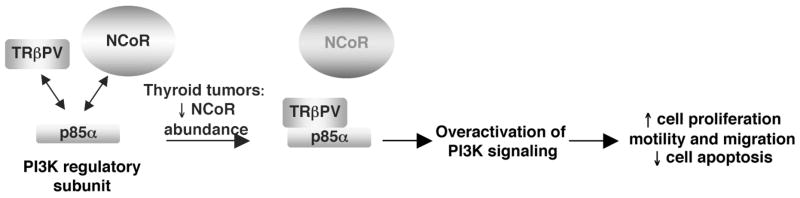
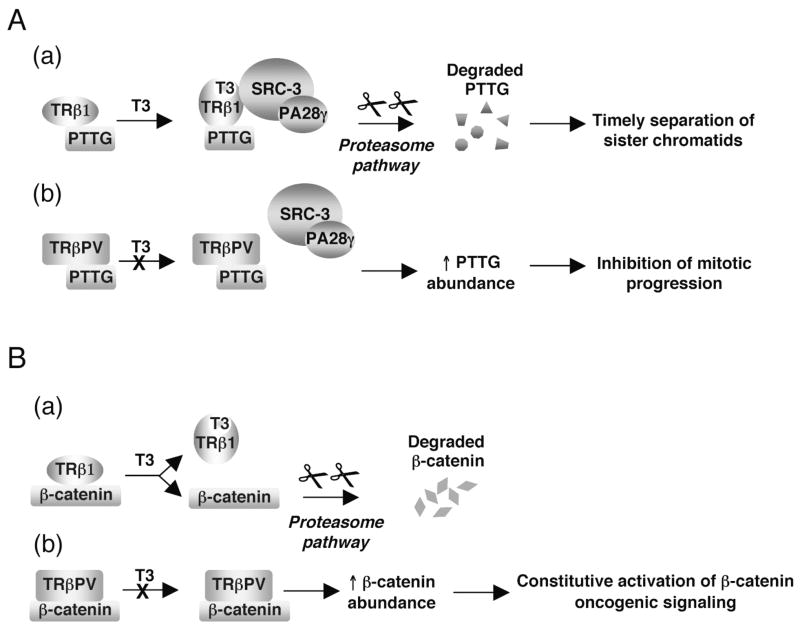
The thyroid hormone receptors (TRs) are transcription factors that mediate the pleiotropic activities of the thyroid hormone, T3. Four T3-binding isoforms, TRalpha1, TRbeta1, TRbeta2, and TRbeta3, are encoded by two genes, THRA and THRB. Mutations and altered expression of TRs have been reported in human cancers. A targeted germ-line mutation of the Thrbeta gene in the mouse leads to spontaneous development of follicular thyroid carcinoma (TRbeta(PV/PV) mouse). The TRbetaPV mutant has lost T3-binding activity and displays potent dominant negative activity. The striking phenotype of thyroid cancer exhibited by TRbeta(PV/PV) mice has recently led to the discovery of novel non-genomic actions of TRbetaPV that contribute to thyroid carcinogenesis. These actions involve direct physical interaction of TRbetaPV with cellular proteins, namely the regulatory subunit of the phosphatidylinositol 3-kinase (p85alpha), the pituitary tumor transforming gene (PTTG) and beta-catenin, that are critically involved in cell proliferation, motility, migration, and metastasis. Thus, a TRbeta mutant (TRbetaPV), via a novel mode of non-genomic action, acts as an oncogene in thyroid carcinogenesis.
Figures
References
-
- Adams M, Matthews C, Collingwood TN, Tone Y, Beck-Peccoz P, Chatterjee KK. Genetic analysis of 29 kindreds with generalized and pituitary resistance to thyroid hormone. Identification of thirteen novel mutations in the thyroid hormone receptor beta gene. J Clin Invest. 1994;94:506–15. - PMC - PubMed
-
- Ando S, Sarlis NJ, Krishnan J, Feng X, Refetoff S, Zhang MQ, Oldfield EH, Yen PM. Aberrant alternative splicing of thyroid hormone receptor in a TSH-secreting pituitary tumor is a mechanism for hormone resistance. Mol Endocrinol. 2001a;15:1529–38. - PubMed
-
- Ando S, Sarlis NJ, Oldfield EH, Yen PM. Somatic mutation of TRbeta can cause a defect in negative regulation of TSH in a TSH-secreting pituitary tumor. J Clin Endocrinol Metab. 2001b;86:5572–6. - PubMed
-
- Bronnegard M, Torring O, Boos J, Sylven C, Marcus C, Wallin G. Expression of thyrotropin receptor and thyroid hormone receptor messenger ribonucleic acid in normal, hyperplastic, and neoplastic human thyroid tissue. J Clin Endocrinol Metab. 1994;79:384–9. - PubMed
-
- Cao X, Kambe F, Moeller LC, Refetoff S, Seo H. Thyroid hormone induces rapid activation of Akt/protein kinase B-mammalian target of rapamycin-p70S6K cascade through phosphatidylinositol 3-kinase in human fibroblasts. Mol Endocrinol. 2005;19:102–12. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
