

Bile acid-induced epidermal growth factor receptor activation in quiescent rat hepatic stellate cells can trigger both proliferation and apoptosis
- PMID: 19553664
- PMCID: PMC2755942
- DOI: 10.1074/jbc.M109.005355
Bile acid-induced epidermal growth factor receptor activation in quiescent rat hepatic stellate cells can trigger both proliferation and apoptosis
Abstract
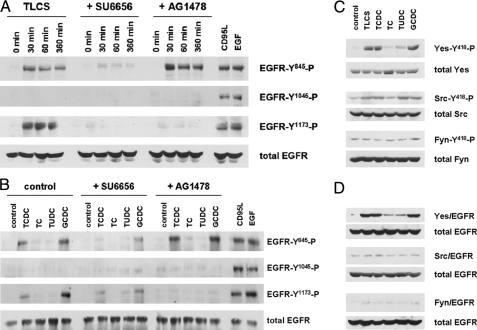
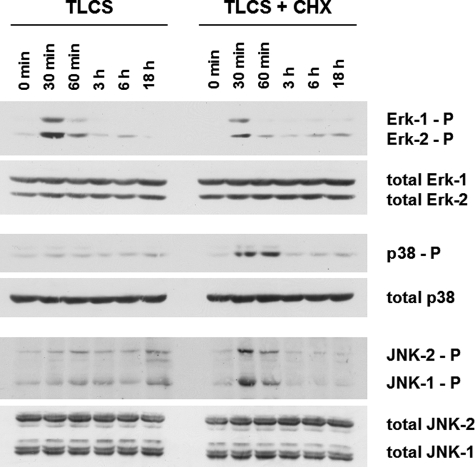
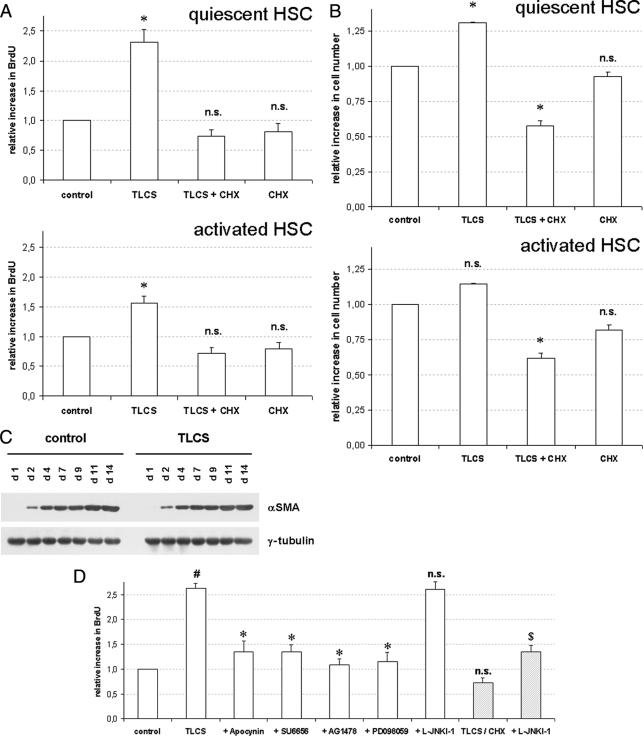
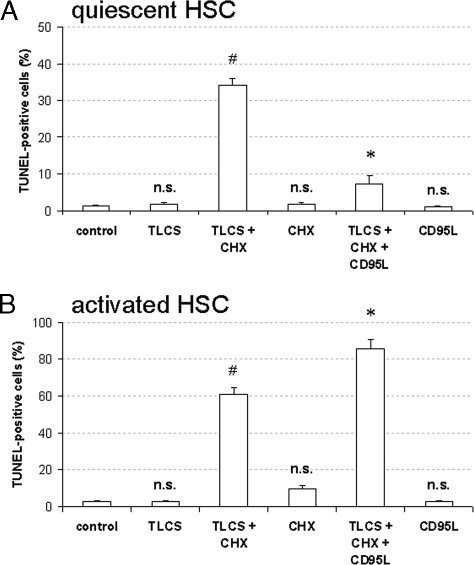
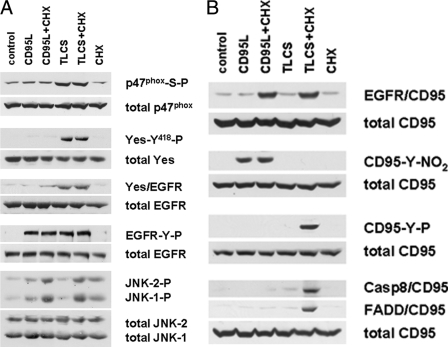
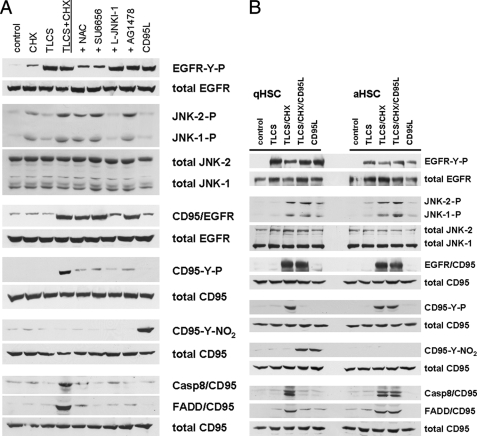
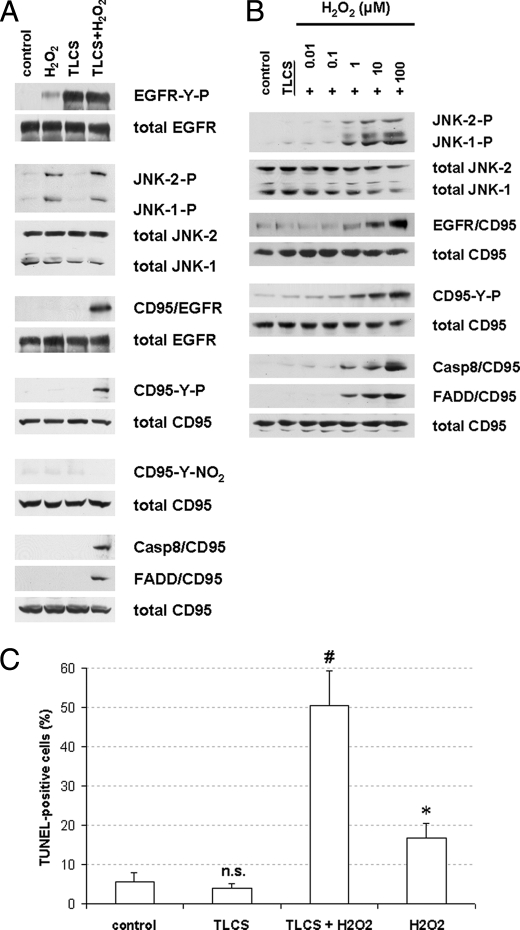
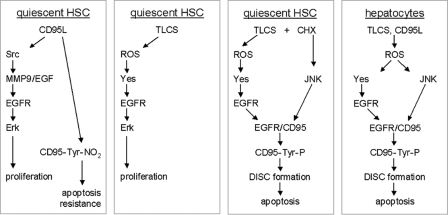
Bile acids have been reported to induce epidermal growth factor receptor (EGFR) activation and subsequent proliferation of activated hepatic stellate cells (HSC), but the underlying mechanisms and whether quiescent HSC are also a target for bile acid-induced proliferation or apoptosis remained unclear. Therefore, primary rat HSC were cultured for up to 48 h and analyzed for their proliferative/apoptotic responses toward bile acids. Hydrophobic bile acids, i.e. taurolithocholate 3-sulfate, taurochenodeoxycholate, and glycochenodeoxycholate, but not taurocholate or tauroursodeoxycholate, induced Yes-dependent EGFR phosphorylation. Simultaneously, hydrophobic bile acids induced phosphorylation of the NADPH oxidase subunit p47(phox) and formation of reactive oxygen species (ROS). ROS production was sensitive to inhibition of acidic sphingomyelinase, protein kinase Czeta, and NADPH oxidases. All maneuvers which prevented bile acid-induced ROS formation also prevented Yes and subsequent EGFR phosphorylation. Taurolithocholate 3-sulfate-induced EGFR activation was followed by extracellular signal-regulated kinase 1/2, but not c-Jun N-terminal kinase (JNK) activation, and stimulated HSC proliferation. When, however, a JNK signal was induced by coadministration of cycloheximide or hydrogen peroxide (H2O2), activated EGFR associated with CD95 and triggered EGFR-mediated CD95-tyrosine phosphorylation and subsequent formation of the death-inducing signaling complex. In conclusion, hydrophobic bile acids lead to a NADPH oxidase-driven ROS generation followed by a Yes-mediated EGFR activation in quiescent primary rat HSC. This proliferative signal shifts to an apoptotic signal when a JNK signal simultaneously comes into play.
Figures


Similar articles
-
Bile salt-induced hepatocyte apoptosis involves epidermal growth factor receptor-dependent CD95 tyrosine phosphorylation.Gastroenterology. 2003 Sep;125(3):839-53. doi: 10.1016/s0016-5085(03)01055-2. Gastroenterology. 2003. PMID: 12949729
-
Involvement of the Src family kinase yes in bile salt-induced apoptosis.Gastroenterology. 2004 Nov;127(5):1540-57. doi: 10.1053/j.gastro.2004.08.056. Gastroenterology. 2004. PMID: 15521021
-
Bile salt-induced apoptosis involves NADPH oxidase isoform activation.Gastroenterology. 2005 Dec;129(6):2009-31. doi: 10.1053/j.gastro.2005.09.023. Gastroenterology. 2005. PMID: 16344068
-
Hyperosmotic activation of the CD95 system.Methods Enzymol. 2007;428:145-60. doi: 10.1016/S0076-6879(07)28008-5. Methods Enzymol. 2007. PMID: 17875416 Review.
-
Decoding cell death signals in liver inflammation.J Hepatol. 2013 Sep;59(3):583-94. doi: 10.1016/j.jhep.2013.03.033. Epub 2013 Apr 6. J Hepatol. 2013. PMID: 23567086 Review.
Cited by
-
Glycochenodeoxycholate Promotes Liver Fibrosis in Mice with Hepatocellular Cholestasis.Cells. 2020 Jan 23;9(2):281. doi: 10.3390/cells9020281. Cells. 2020. PMID: 31979271 Free PMC article.
-
Protective effect of bile acids on the onset of fructose-induced hepatic steatosis in mice.J Lipid Res. 2010 Dec;51(12):3414-24. doi: 10.1194/jlr.M007179. Epub 2010 Sep 16. J Lipid Res. 2010. PMID: 20847296 Free PMC article.
-
Exposure of Barrett's and esophageal adenocarcinoma cells to bile acids activates EGFR-STAT3 signaling axis via induction of APE1.Oncogene. 2018 Nov;37(46):6011-6024. doi: 10.1038/s41388-018-0388-8. Epub 2018 Jul 10. Oncogene. 2018. PMID: 29991802 Free PMC article.
-
Uncovering a Novel Functional Interaction Between Adult Hepatic Progenitor Cells, Inflammation and EGFR Signaling During Bile Acids-Induced Injury.Int J Biol Sci. 2024 Apr 8;20(7):2339-2355. doi: 10.7150/ijbs.90645. eCollection 2024. Int J Biol Sci. 2024. PMID: 38725853 Free PMC article.
-
EGFR Signaling in Liver Diseases.Int J Mol Sci. 2015 Dec 29;17(1):30. doi: 10.3390/ijms17010030. Int J Mol Sci. 2015. PMID: 26729094 Free PMC article. Review.
References
-
- Chieco P., Romagnoli E., Aicardi G., Suozzi A., Forti G. C., Roda A. (1997) Histochem. J. 29, 875–883 - PubMed
-
- Miyoshi H., Rust C., Roberts P. J., Burgart L. J., Gores G. J. (1999) Gastroenterology 117, 669–677 - PubMed
-
- Yerushalmi B., Dahl R., Devereaux M. W., Gumpricht E., Sokol R. J. (2001) Hepatology 33, 616–626 - PubMed
-
- Graf D., Kurz A. K., Fischer R., Reinehr R., Häussinger D. (2002) Gastroenterology 122, 1411–1427 - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
