

Carnitine insufficiency caused by aging and overnutrition compromises mitochondrial performance and metabolic control
- PMID: 19553674
- PMCID: PMC2755692
- DOI: 10.1074/jbc.M109.032888
Carnitine insufficiency caused by aging and overnutrition compromises mitochondrial performance and metabolic control
Abstract
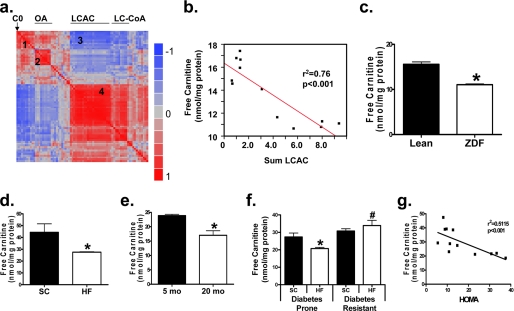
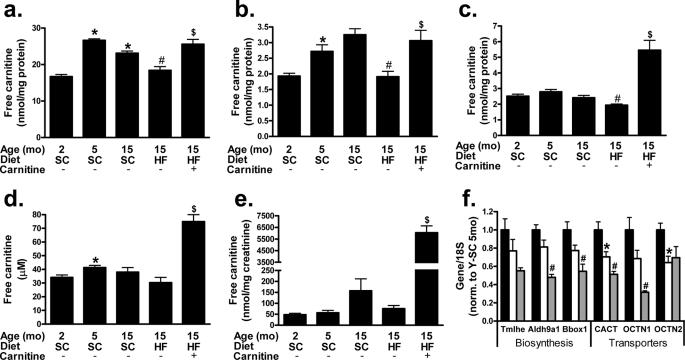
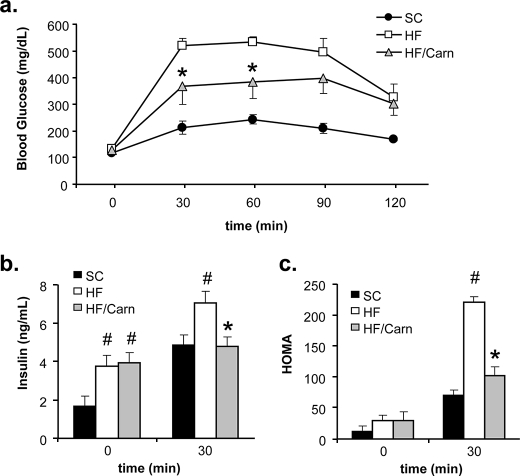
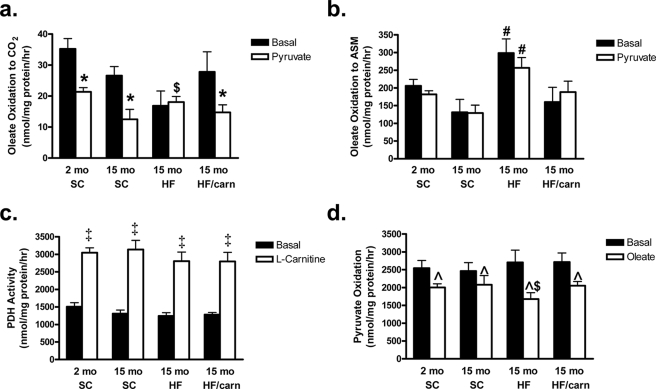
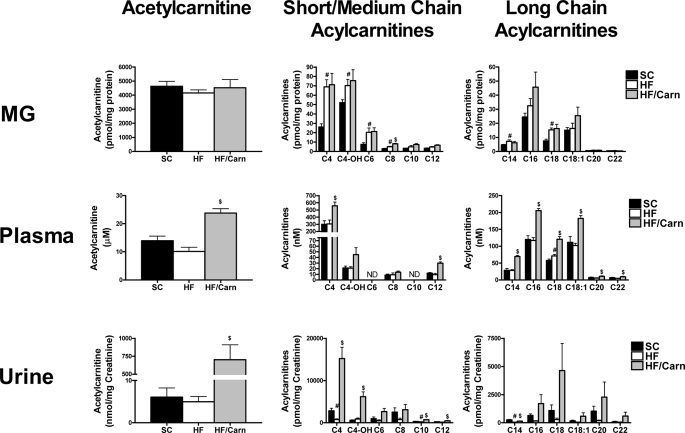
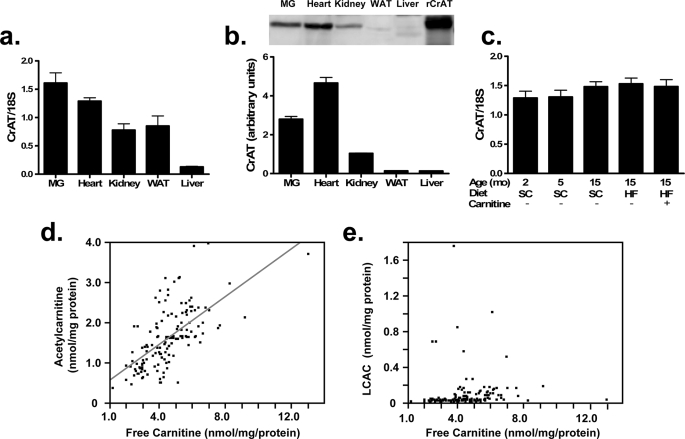
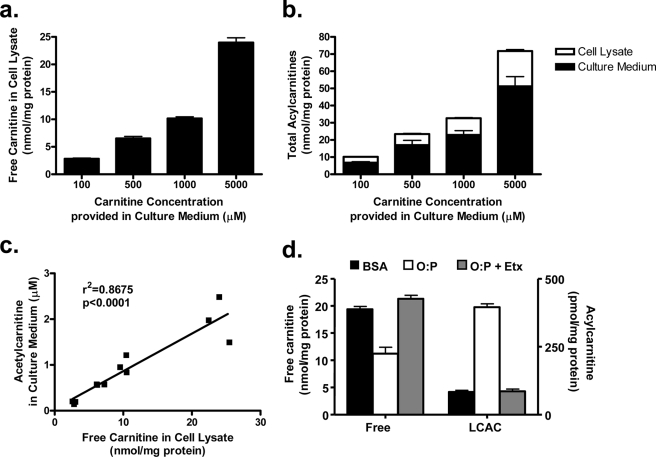
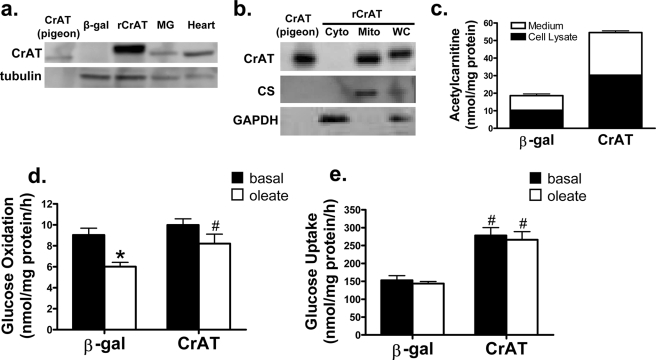
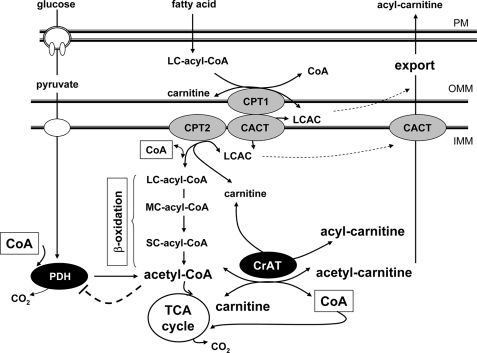
In addition to its essential role in permitting mitochondrial import and oxidation of long chain fatty acids, carnitine also functions as an acyl group acceptor that facilitates mitochondrial export of excess carbons in the form of acylcarnitines. Recent evidence suggests carnitine requirements increase under conditions of sustained metabolic stress. Accordingly, we hypothesized that carnitine insufficiency might contribute to mitochondrial dysfunction and obesity-related impairments in glucose tolerance. Consistent with this prediction whole body carnitine diminution was identified as a common feature of insulin-resistant states such as advanced age, genetic diabetes, and diet-induced obesity. In rodents fed a lifelong (12 month) high fat diet, compromised carnitine status corresponded with increased skeletal muscle accumulation of acylcarnitine esters and diminished hepatic expression of carnitine biosynthetic genes. Diminished carnitine reserves in muscle of obese rats was accompanied by marked perturbations in mitochondrial fuel metabolism, including low rates of complete fatty acid oxidation, elevated incomplete beta-oxidation, and impaired substrate switching from fatty acid to pyruvate. These mitochondrial abnormalities were reversed by 8 weeks of oral carnitine supplementation, in concert with increased tissue efflux and urinary excretion of acetylcarnitine and improvement of whole body glucose tolerance. Acetylcarnitine is produced by the mitochondrial matrix enzyme, carnitine acetyltransferase (CrAT). A role for this enzyme in combating glucose intolerance was further supported by the finding that CrAT overexpression in primary human skeletal myocytes increased glucose uptake and attenuated lipid-induced suppression of glucose oxidation. These results implicate carnitine insufficiency and reduced CrAT activity as reversible components of the metabolic syndrome.
Figures
Similar articles
-
Obesity and lipid stress inhibit carnitine acetyltransferase activity.J Lipid Res. 2014 Apr;55(4):635-44. doi: 10.1194/jlr.M043448. Epub 2014 Jan 6. J Lipid Res. 2014. PMID: 24395925 Free PMC article.
-
Carnitine supplementation in high-fat diet-fed rats does not ameliorate lipid-induced skeletal muscle mitochondrial dysfunction in vivo.Am J Physiol Endocrinol Metab. 2015 Oct 1;309(7):E670-8. doi: 10.1152/ajpendo.00144.2015. Epub 2015 Aug 18. Am J Physiol Endocrinol Metab. 2015. PMID: 26286868
-
Carnitine supplementation alleviates lipid metabolism derangements and protects against oxidative stress in non-obese hereditary hypertriglyceridemic rats.Appl Physiol Nutr Metab. 2015 Mar;40(3):280-91. doi: 10.1139/apnm-2014-0163. Appl Physiol Nutr Metab. 2015. PMID: 25723909
-
Carnitine O-Acetyltransferase as a Central Player in Lipid and Branched-Chain Amino Acid Metabolism, Epigenetics, Cell Plasticity, and Organelle Function.Biomolecules. 2025 Feb 2;15(2):216. doi: 10.3390/biom15020216. Biomolecules. 2025. PMID: 40001519 Free PMC article. Review.
-
Regulation by carnitine of myocardial fatty acid and carbohydrate metabolism under normal and pathological conditions.Basic Res Cardiol. 2000 Apr;95(2):75-83. doi: 10.1007/s003950050167. Basic Res Cardiol. 2000. PMID: 10826498 Review.
Cited by
-
P38α MAPK Coordinates Mitochondrial Adaptation to Caloric Surplus in Skeletal Muscle.Int J Mol Sci. 2024 Jul 16;25(14):7789. doi: 10.3390/ijms25147789. Int J Mol Sci. 2024. PMID: 39063031 Free PMC article.
-
Dipeptidyl peptidase-4 inhibition ameliorates Western diet-induced hepatic steatosis and insulin resistance through hepatic lipid remodeling and modulation of hepatic mitochondrial function.Diabetes. 2015 Jun;64(6):1988-2001. doi: 10.2337/db14-0804. Epub 2015 Jan 20. Diabetes. 2015. PMID: 25605806 Free PMC article.
-
Aerobic capacity and hepatic mitochondrial lipid oxidation alters susceptibility for chronic high-fat diet-induced hepatic steatosis.Am J Physiol Endocrinol Metab. 2016 Oct 1;311(4):E749-E760. doi: 10.1152/ajpendo.00178.2016. Epub 2016 Sep 6. Am J Physiol Endocrinol Metab. 2016. PMID: 27600823 Free PMC article.
-
Muscle-specific deletion of carnitine acetyltransferase compromises glucose tolerance and metabolic flexibility.Cell Metab. 2012 May 2;15(5):764-77. doi: 10.1016/j.cmet.2012.04.005. Cell Metab. 2012. PMID: 22560225 Free PMC article.
-
Efficacy of Dietary Supplements to Reduce Liver Fat.Nutrients. 2020 Jul 31;12(8):2302. doi: 10.3390/nu12082302. Nutrients. 2020. PMID: 32751906 Free PMC article.
References
-
- Kelley D. E., Goodpaster B., Wing R. R., Simoneau J. A. (1999) Am. J. Physiol. 277,E1130–E1141 - PubMed
-
- Schrauwen P., Hesselink M. K. (2004) Diabetes 53,1412–1417 - PubMed
-
- Koves T. R., Li P., An J., Akimoto T., Slentz D., Ilkayeva O., Dohm G. L., Yan Z., Newgard C. B., Muoio D. M. (2005) J. Biol. Chem. 280,33588–33598 - PubMed
-
- Koves T. R., Ussher J. R., Noland R. C., Slentz D., Mosedale M., Ilkayeva O., Bain J., Stevens R., Dyck J. R., Newgard C. B., Lopaschuk G. D., Muoio D. M. (2008) Cell Metab. 7,45–56 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
