

Advances in light microscopy for neuroscience
- PMID: 19555292
- PMCID: PMC2820375
- DOI: 10.1146/annurev.neuro.051508.135540
Advances in light microscopy for neuroscience
Abstract
Since the work of Golgi and Cajal, light microscopy has remained a key tool for neuroscientists to observe cellular properties. Ongoing advances have enabled new experimental capabilities using light to inspect the nervous system across multiple spatial scales, including ultrastructural scales finer than the optical diffraction limit. Other progress permits functional imaging at faster speeds, at greater depths in brain tissue, and over larger tissue volumes than previously possible. Portable, miniaturized fluorescence microscopes now allow brain imaging in freely behaving mice. Complementary progress on animal preparations has enabled imaging in head-restrained behaving animals, as well as time-lapse microscopy studies in the brains of live subjects. Mouse genetic approaches permit mosaic and inducible fluorescence-labeling strategies, whereas intrinsic contrast mechanisms allow in vivo imaging of animals and humans without use of exogenous markers. This review surveys such advances and highlights emerging capabilities of particular interest to neuroscientists.
Figures


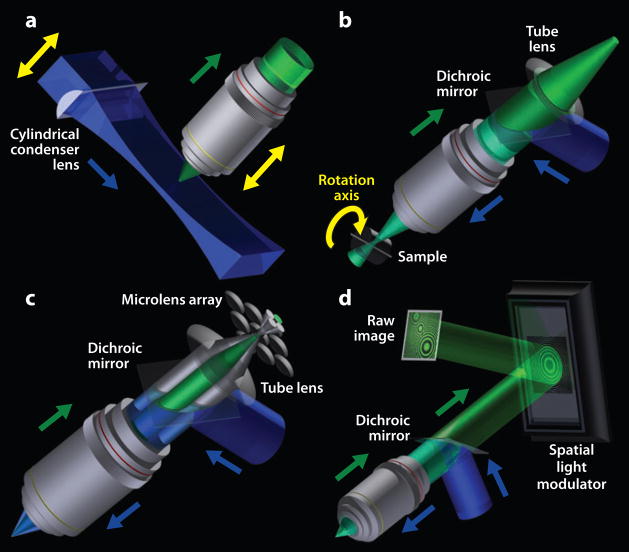






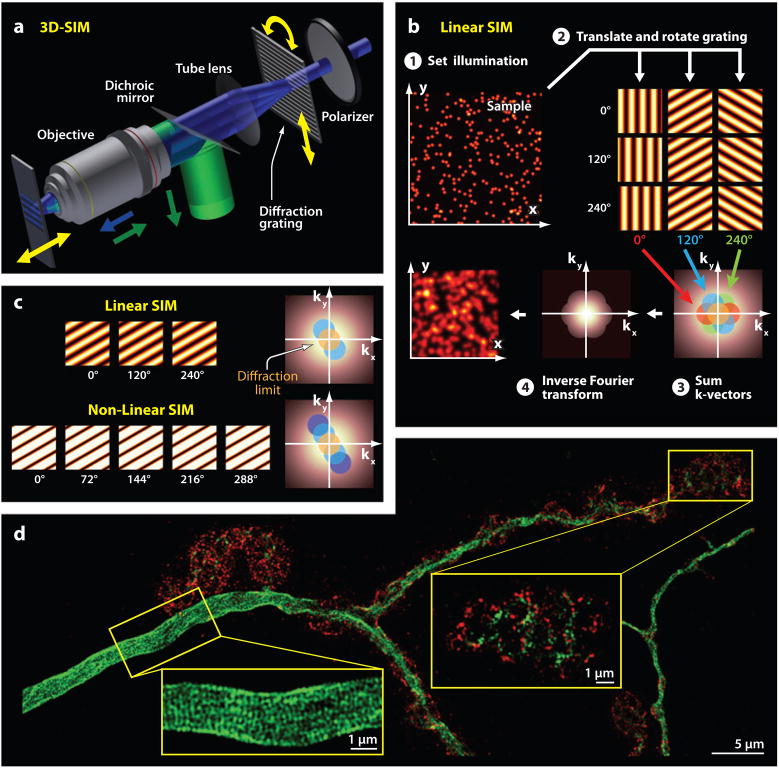
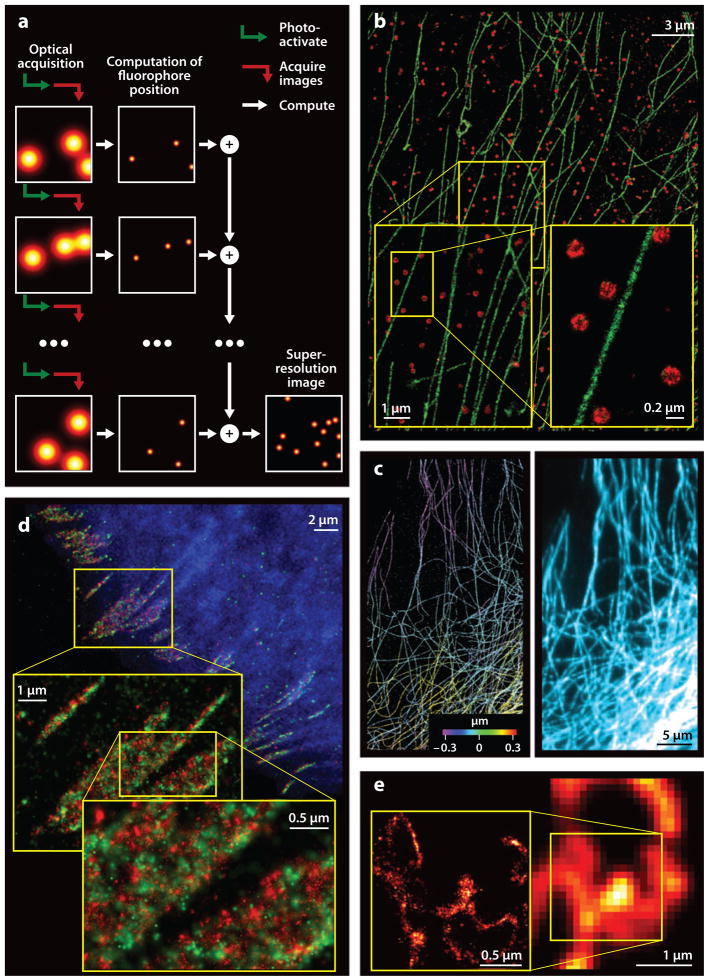

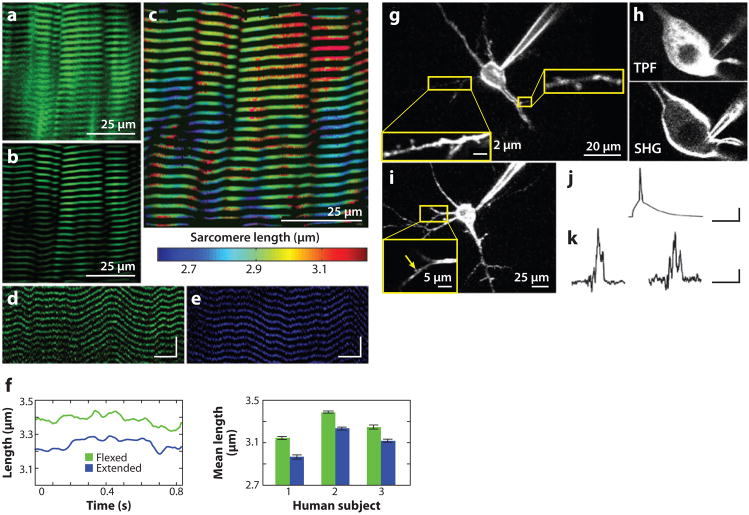


References
-
- Adelsberger H, Garaschuk O, Konnerth A. Cortical calcium waves in resting newborn mice. Nat Neurosci. 2005;8:988–90. - PubMed
-
- Aguirre AD, Nishizawa N, Fujimoto JG, Seitz W, Lederer M, Kopf D. Continuum generation in a novel photonic crystal fiber for ultrahigh resolution optical coherence tomography at 800 nm and 1300 nm. Opt Express. 2006b;14:1145–60. - PubMed
-
- Akkin T, Davé DP, Milner TE, Rylander HG. Detection of neural activity using phase-sensitive optical low-coherence reflectometry. Opt Express. 2004;12:2377–86. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
