

Mutation in the AP4M1 gene provides a model for neuroaxonal injury in cerebral palsy
- PMID: 19559397
- PMCID: PMC2706965
- DOI: 10.1016/j.ajhg.2009.06.004
Mutation in the AP4M1 gene provides a model for neuroaxonal injury in cerebral palsy
Abstract
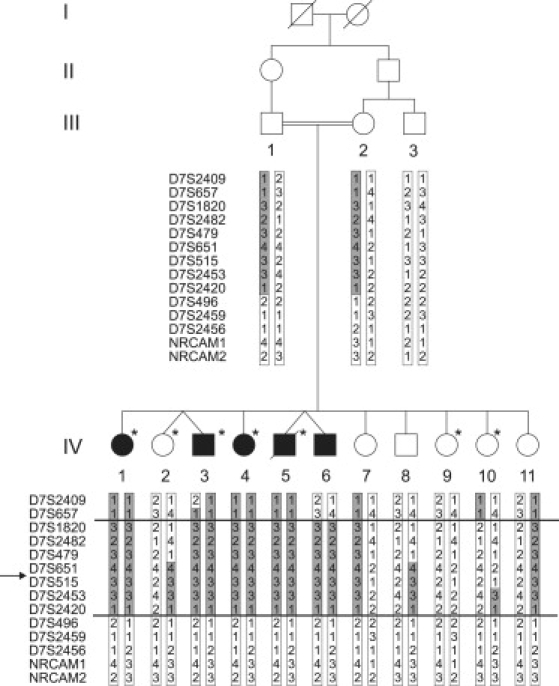
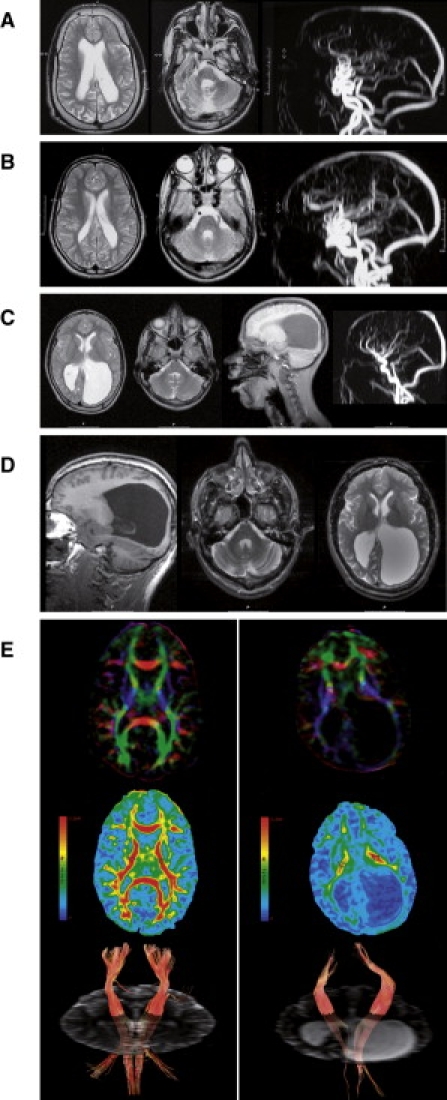
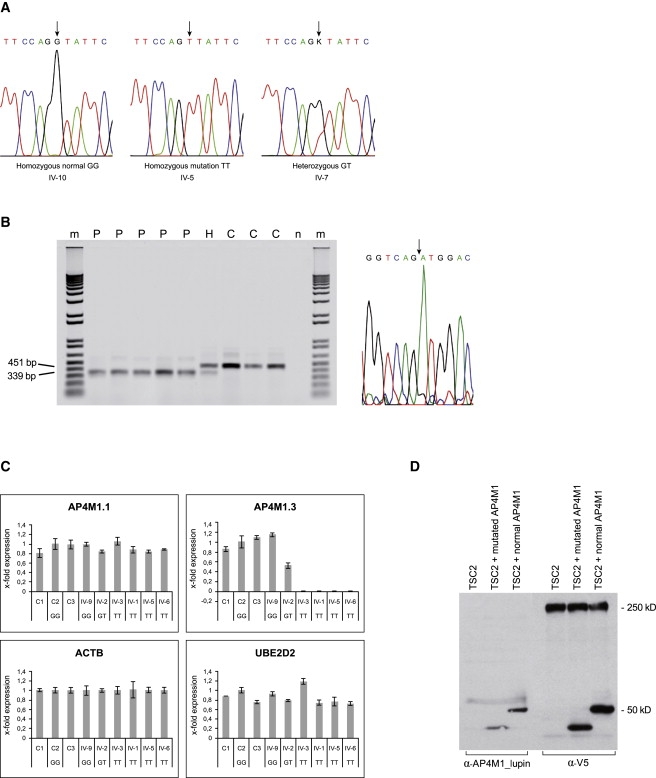
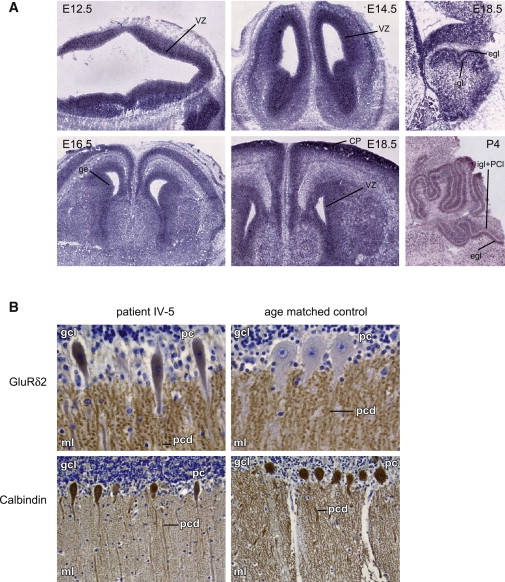
Cerebral palsy due to perinatal injury to cerebral white matter is usually not caused by genetic mutations, but by ischemia and/or inflammation. Here, we describe an autosomal-recessive type of tetraplegic cerebral palsy with mental retardation, reduction of cerebral white matter, and atrophy of the cerebellum in an inbred sibship. The phenotype was recorded and evolution followed for over 20 years. Brain lesions were studied by diffusion tensor MR tractography. Homozygosity mapping with SNPs was performed for identification of the chromosomal locus for the disease. In the 14 Mb candidate region on chromosome 7q22, RNA expression profiling was used for selecting among the 203 genes in the area. In postmortem brain tissue available from one patient, histology and immunohistochemistry were performed. Disease course and imaging were mostly reminiscent of hypoxic-ischemic tetraplegic cerebral palsy, with neuroaxonal degeneration and white matter loss. In all five patients, a donor splice site pathogenic mutation in intron 14 of the AP4M1 gene (c.1137+1G-->T), was identified. AP4M1, encoding for the mu subunit of the adaptor protein complex-4, is involved in intracellular trafficking of glutamate receptors. Aberrant GluRdelta2 glutamate receptor localization and dendritic spine morphology were observed in the postmortem brain specimen. This disease entity, which we refer to as congenital spastic tetraplegia (CST), is therefore a genetic model for congenital cerebral palsy with evidence for neuroaxonal damage and glutamate receptor abnormality, mimicking perinatally acquired hypoxic-ischemic white matter injury.
Figures
References
-
- Rennie J.M., Hagmann C.F., Robertson N.J. Outcome after intrapartum hypoxic ischaemia at term. Semin. Fetal Neonatal Med. 2007;12:398–407. - PubMed
-
- Nelson K.B. Causative factors in cerebral palsy. Clin. Obstet. Gynecol. 2008;51:749–762. - PubMed
-
- Bax M., Tydeman C., Flodmark O. Clinical and MRI correlates of cerebral palsy: The European Cerebral Palsy Study. JAMA. 2006;296:1602–1608. - PubMed
-
- Volpe J.J. Neurobiology of periventricular leukomalacia in the premature infant. Pediatr. Res. 2001;50:553–562. - PubMed
-
- Folkerth R.D. Neuropathologic substrate of cerebral palsy. J. Child Neurol. 2005;20:940–949. - PubMed
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
