

Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads
- PMID: 19561018
- PMCID: PMC2781750
- DOI: 10.1093/bioinformatics/btp394
Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads
Abstract
Motivation: There is a strong demand in the genomic community to develop effective algorithms to reliably identify genomic variants. Indel detection using next-gen data is difficult and identification of long structural variations is extremely challenging.
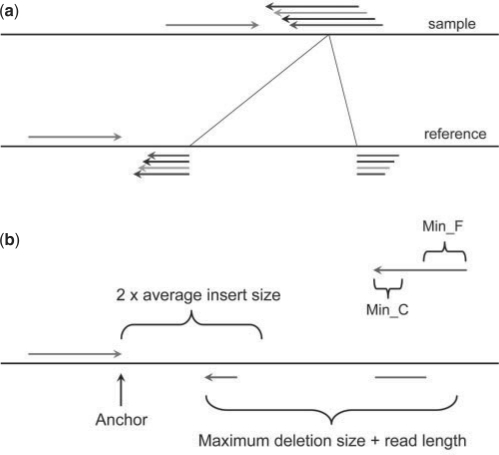
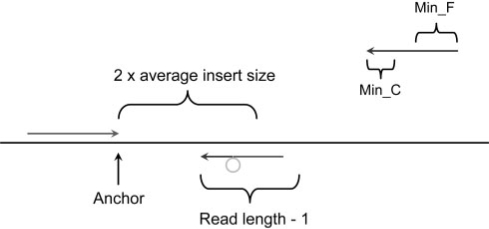
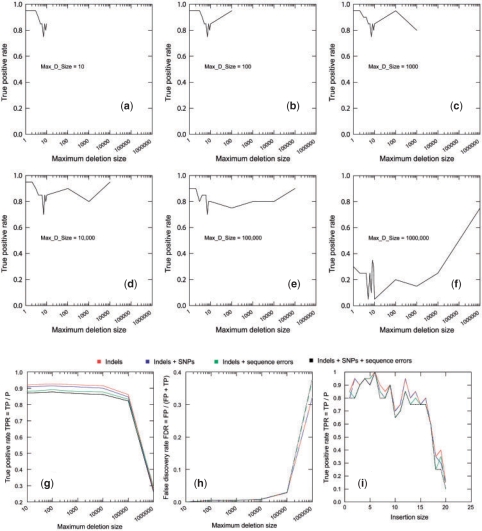
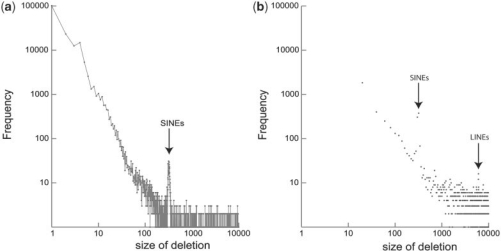
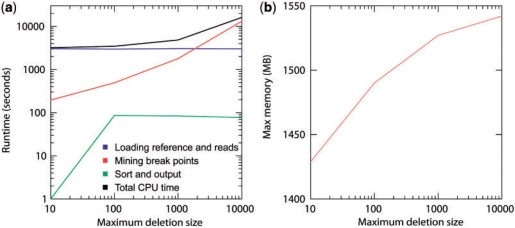
Results: We present Pindel, a pattern growth approach, to detect breakpoints of large deletions and medium-sized insertions from paired-end short reads. We use both simulated reads and real data to demonstrate the efficiency of the computer program and accuracy of the results.
Availability: The binary code and a short user manual can be freely downloaded from http://www.ebi.ac.uk/ approximately kye/pindel/.
Contact: k.ye@lumc.nl; zn1@sanger.ac.uk.
Figures

References
-
- Iafrate AJ, et al. Detection of large-scale variation in the human genome. Nat. Genet. 2004;36:949–951. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
