

High-avidity autoreactive CD4+ T cells induce host CTL, overcome T(regs) and mediate tumor destruction
- PMID: 19561540
- PMCID: PMC2747815
- DOI: 10.1097/CJI.0b013e3181ab1824
High-avidity autoreactive CD4+ T cells induce host CTL, overcome T(regs) and mediate tumor destruction
Abstract
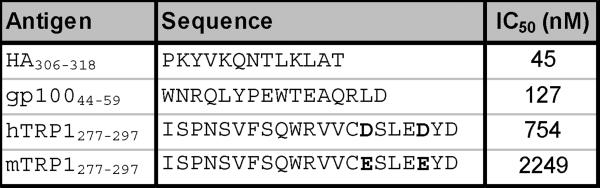
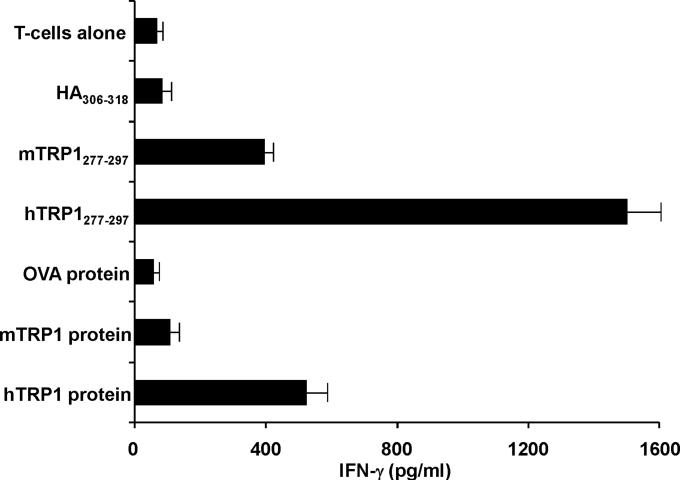
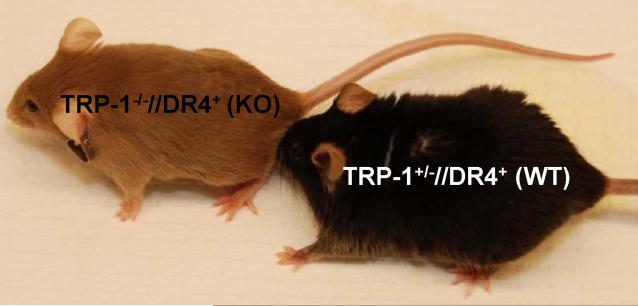
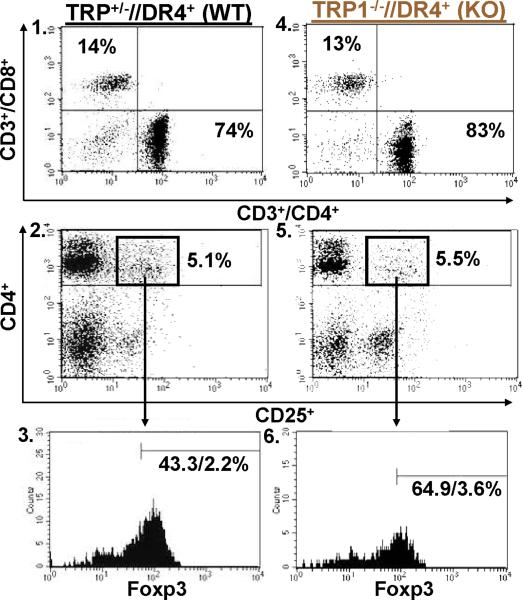
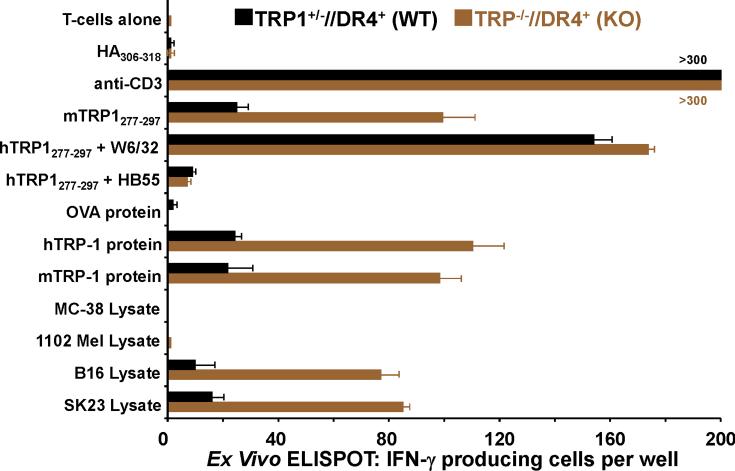
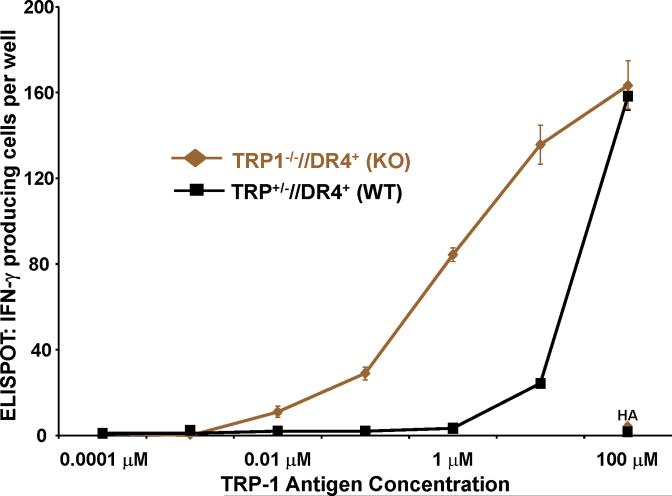
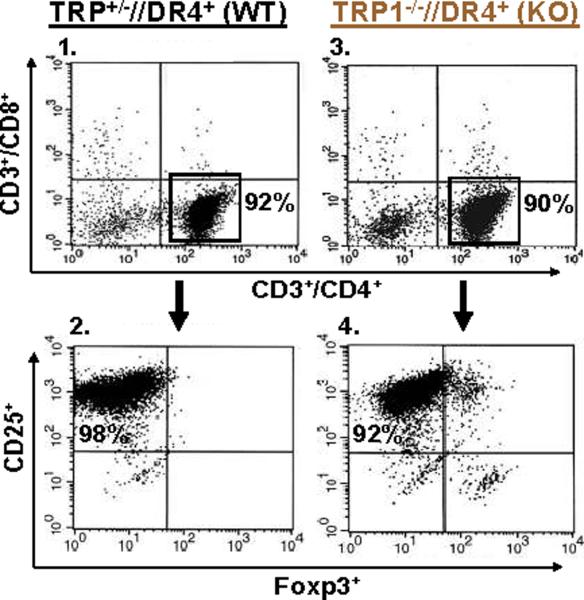
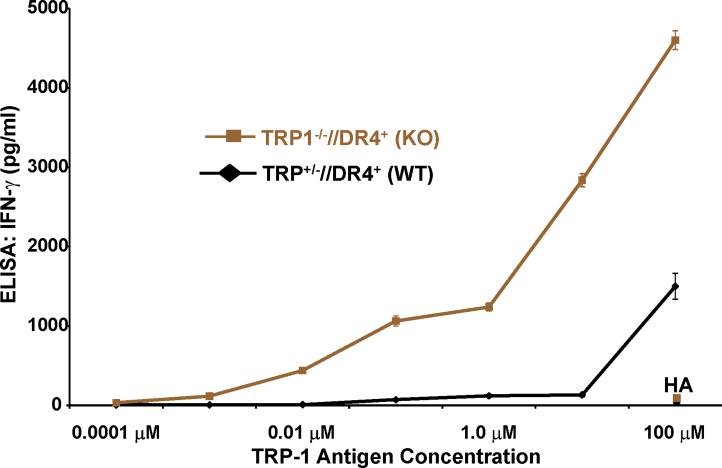
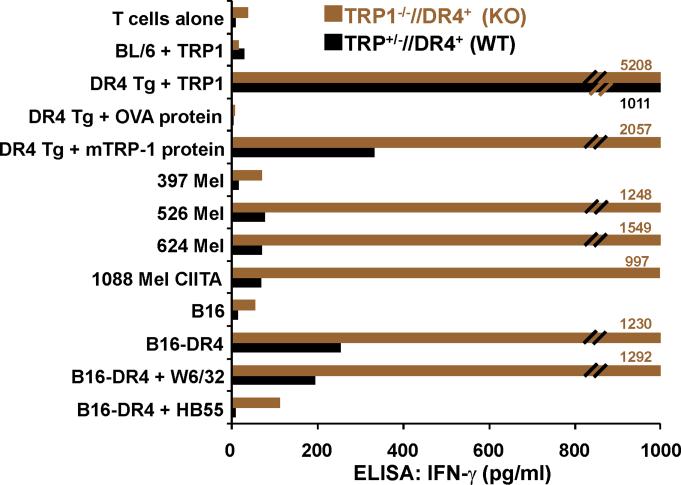
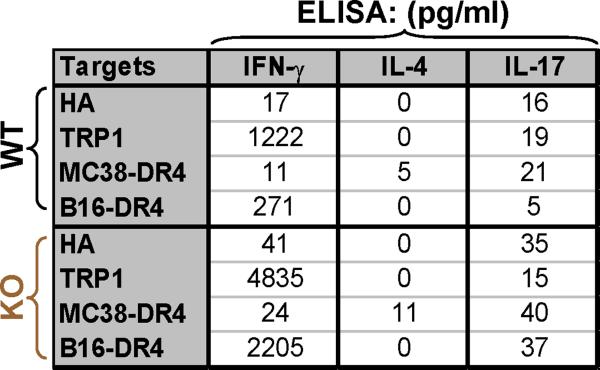
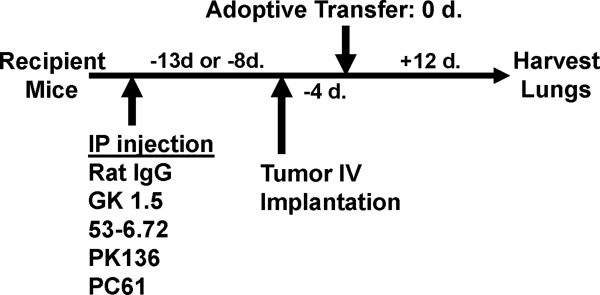
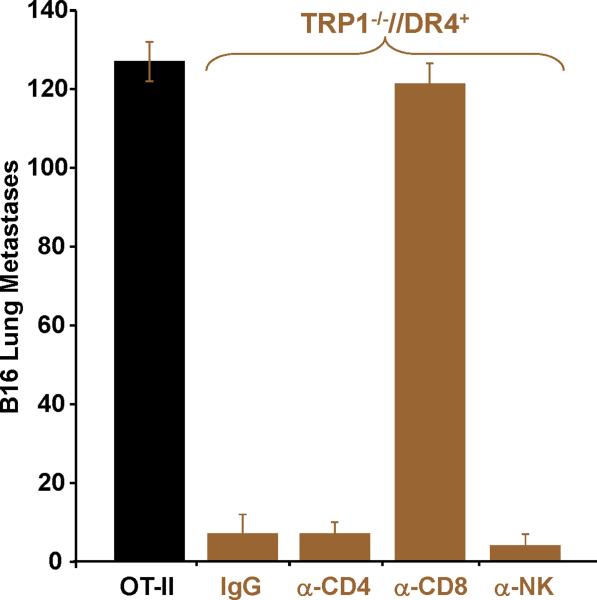
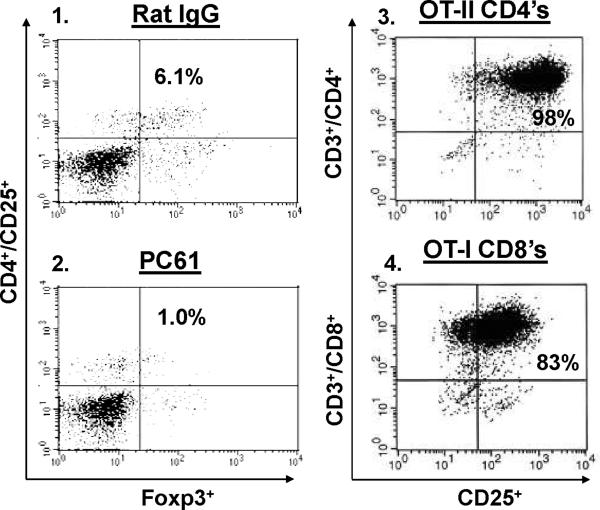
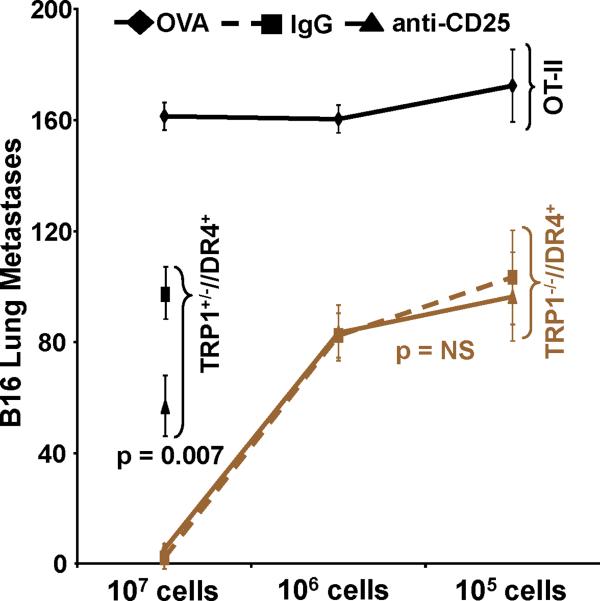
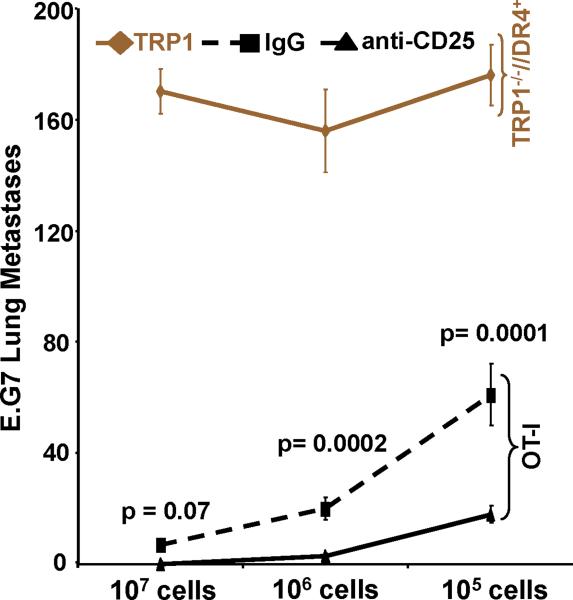
Despite progress made over the past 25 years, existing immunotherapies have limited clinical effectiveness in patients with cancer. Immune tolerance consistently blunts the generated immune response, and the largely solitary focus on CD8+ T cell immunity has proven ineffective in the absence of CD4+ T cell help. To address these twin-tier deficiencies, we developed a translational model of melanoma immunotherapy focused on the exploitation of high-avidity CD4+ T cells that become generated in germline antigen-deficient mice. We had previously identified a tyrosinase-related protein-1 specific HLA-DRB1*0401-restricted epitope. Using this epitope in conjunction with a newly described tyrosinase-related protein-1 germline-knockout, we demonstrate that endogenous tyrosinase-related protein-1 expression alters the functionality of the autoreactive T cell repertoire. More importantly, we show, by using major histocompatibility complex-mismatched combinations, that CD4+ T cells derived from the self-antigen deficient host indirectly triggers the eradication of established B16 lung metastases. We demonstrate that the treatment effect is mediated entirely by endogenous CD8+ T cells and is not affected by the depletion of host regulatory T cells. These findings suggest that high-avidity CD4+ T cells can overcome endogenous conditions and mediate their antitumor effects exclusively through the elicitation of CD8+ T cell immunity.
Figures


References
-
- Boon T, Coulie PG, Van Den Eynde BJ, van der BP. Human T cell responses against melanoma. Annu.Rev.Immunol. 2006;24:175–208. - PubMed
-
- Peggs KS, Segal NH, Allison JP. Targeting immunosupportive cancer therapies: accentuate the positive, eliminate the negative. Cancer Cell. 2007;12:192–199. - PubMed
-
- Lizee G, Cantu MA, Hwu P. Less yin, more yang: confronting the barriers to cancer immunotherapy. Clin.Cancer Res. 2007;13:5250–5255. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
