

Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair
- PMID: 19561612
- PMCID: PMC2730354
- DOI: 10.1038/ni.1758
Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair
Abstract
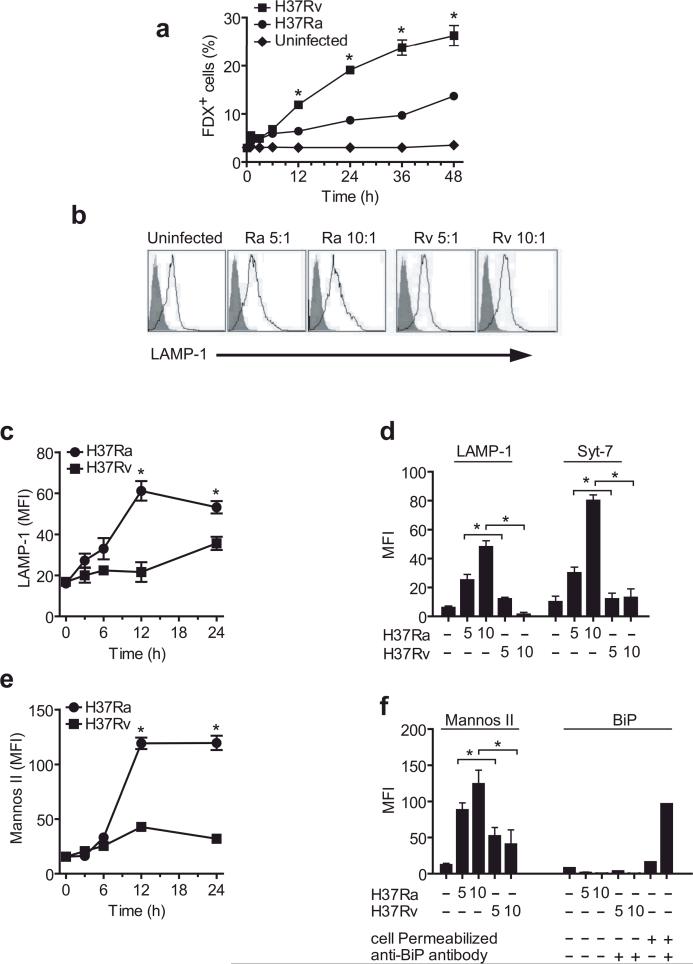
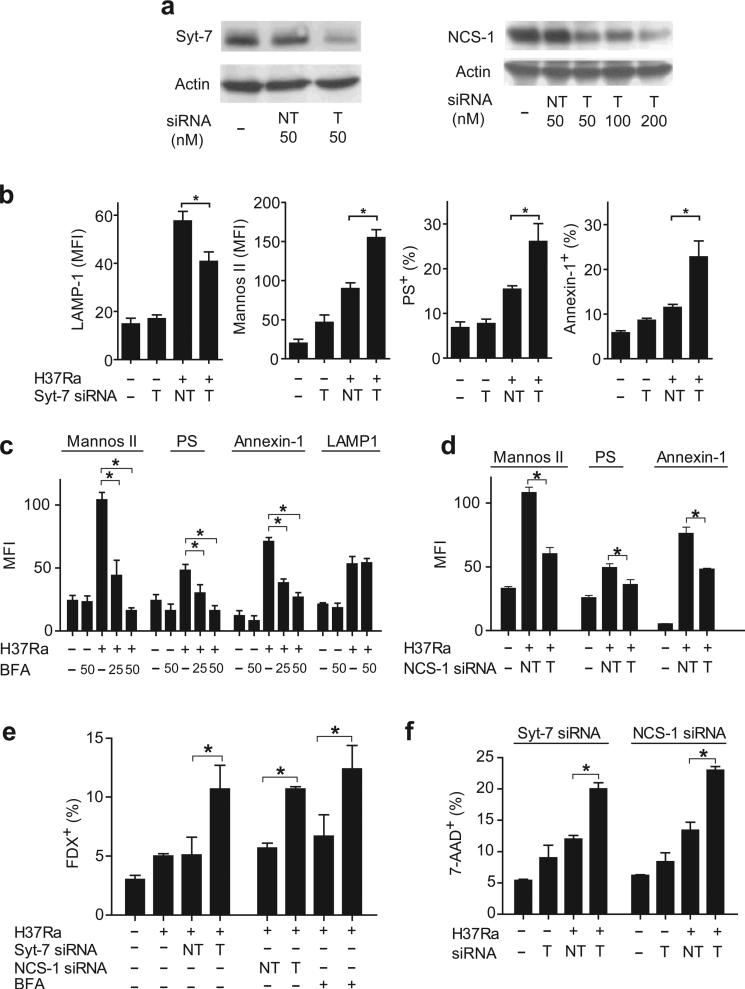
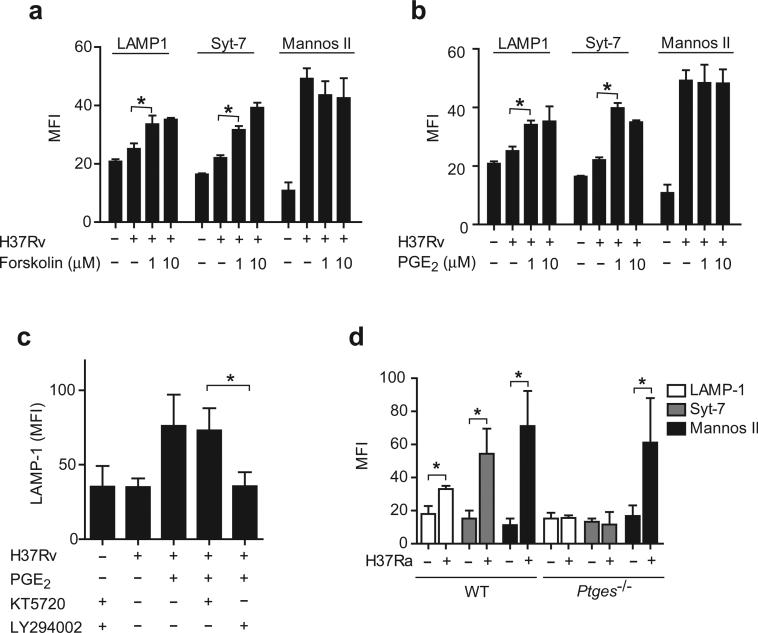
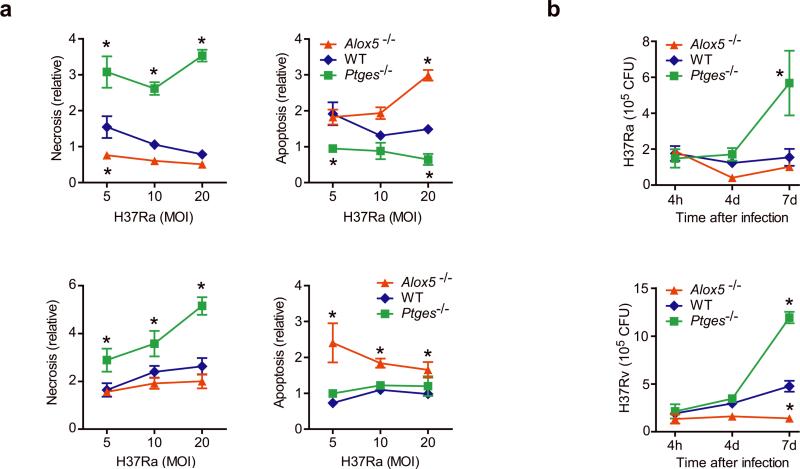
Induction of macrophage necrosis is a strategy used by virulent Mycobacterium tuberculosis (Mtb) to avoid innate host defense. In contrast, attenuated Mtb causes apoptosis, which limits bacterial replication and promotes T cell cross-priming by antigen-presenting cells. Here we show that Mtb infection causes plasma membrane microdisruptions. Resealing of these lesions, a process crucial for preventing necrosis and promoting apoptosis, required translocation of lysosomal and Golgi apparatus-derived vesicles to the plasma membrane. Plasma membrane repair depended on prostaglandin E(2) (PGE(2)), which regulates synaptotagmin 7 (Syt-7), the calcium sensor involved in the lysosome-mediated repair mechanism. By inducing production of lipoxin A(4) (LXA(4)), which blocks PGE(2) biosynthesis, virulent Mtb prevented membrane repair and induced necrosis. Thus, virulent Mtb impairs macrophage plasma membrane repair to evade host defenses.
Figures
References
-
- McNeil PL, Steinhardt RA. Plasma membrane disruption: repair, prevention, adaptation. Annu. Rev. Cell Dev. Biol. 2003;19:697–731. - PubMed
-
- Roy D, et al. A process for controlling intracellular bacterial infections induced by membrane injury. Science. 2004;304:1515–1518. - PubMed
-
- Togo T, Alderton JM, Bi GQ, Steinhardt RA. The mechanism of facilitated cell membrane resealing. J. Cell Sci. 1999;112(Pt 5):719–731. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
