

A mutation in Irak2c identifies IRAK-2 as a central component of the TLR regulatory network of wild-derived mice
- PMID: 19564352
- PMCID: PMC2715079
- DOI: 10.1084/jem.20090490
A mutation in Irak2c identifies IRAK-2 as a central component of the TLR regulatory network of wild-derived mice
Abstract
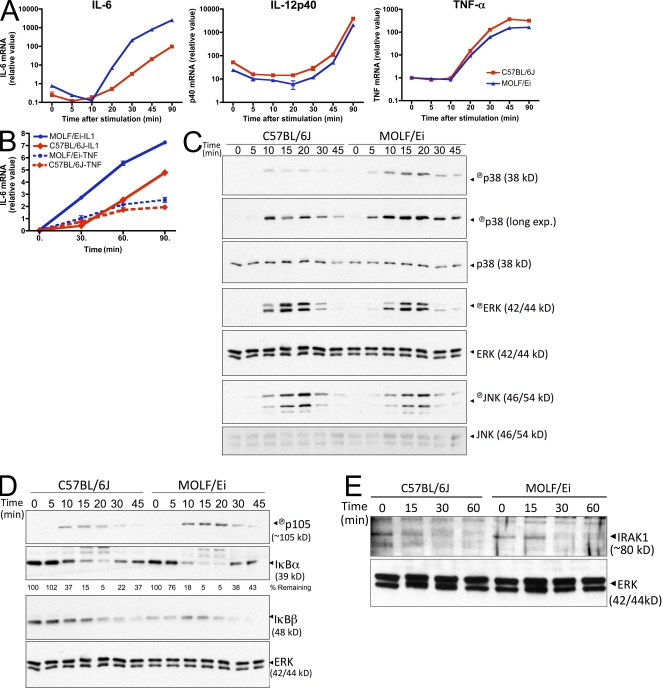
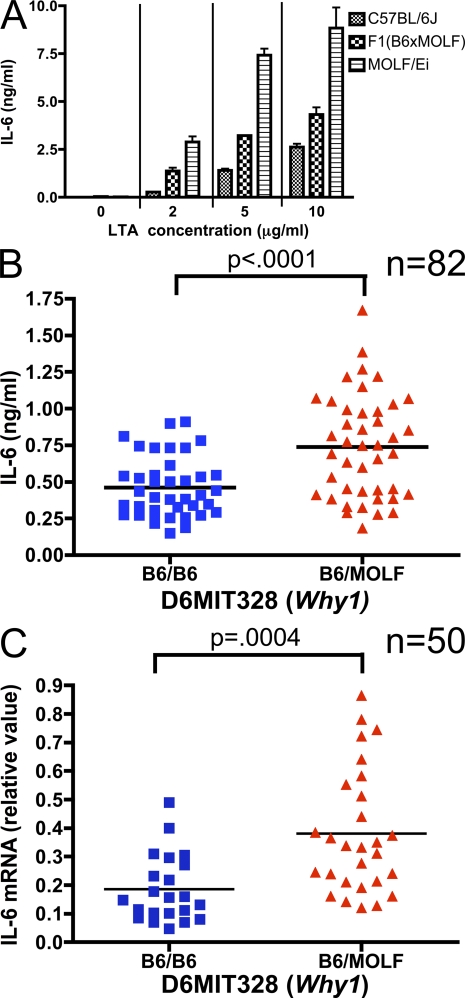
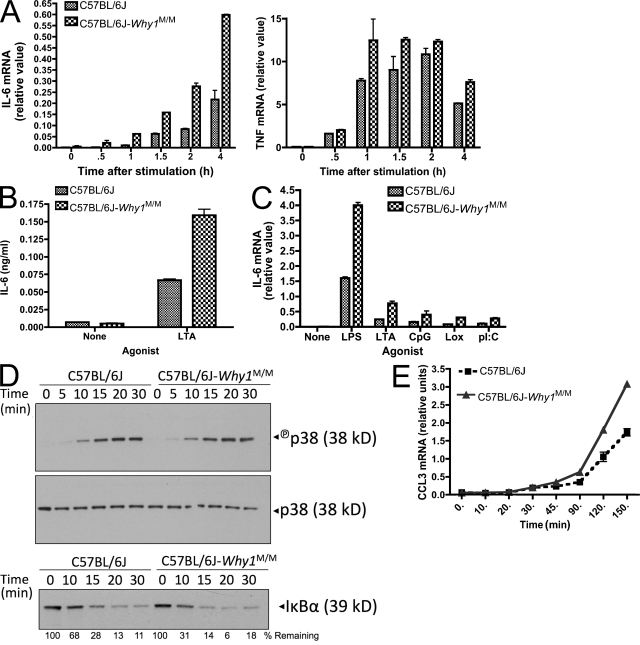
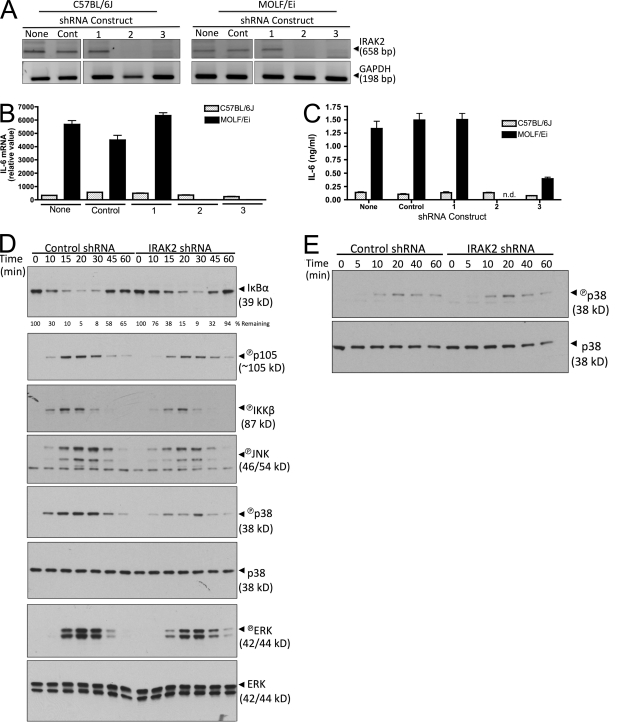
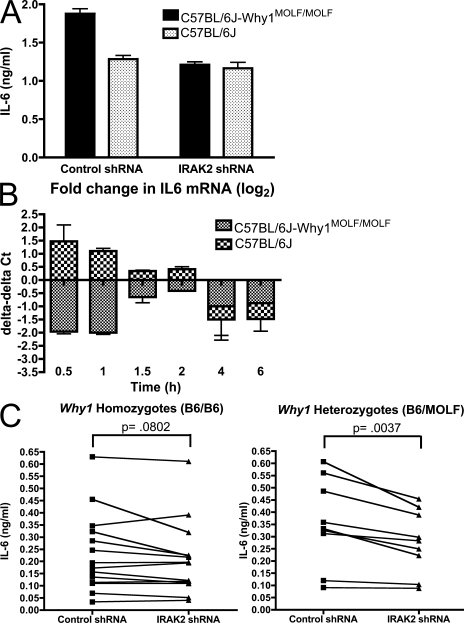
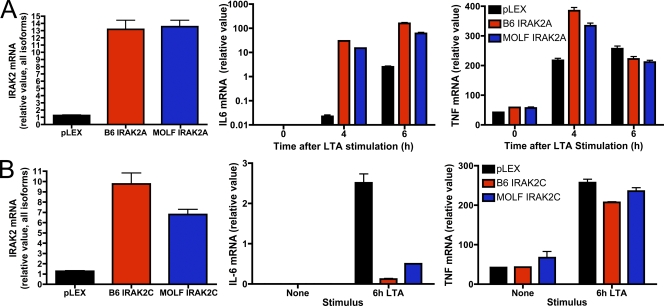
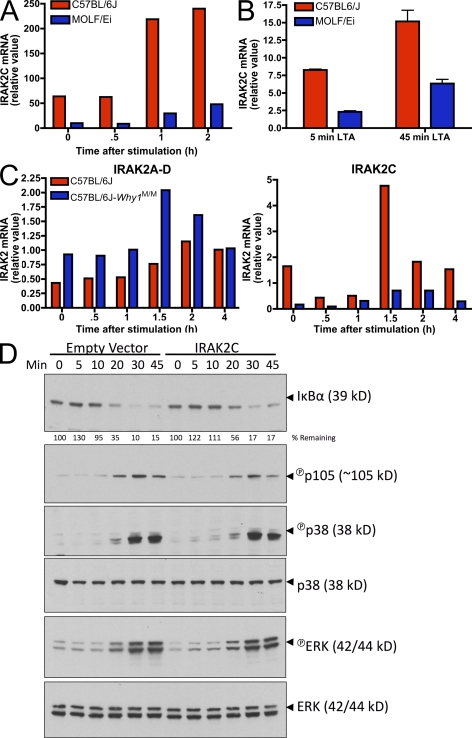
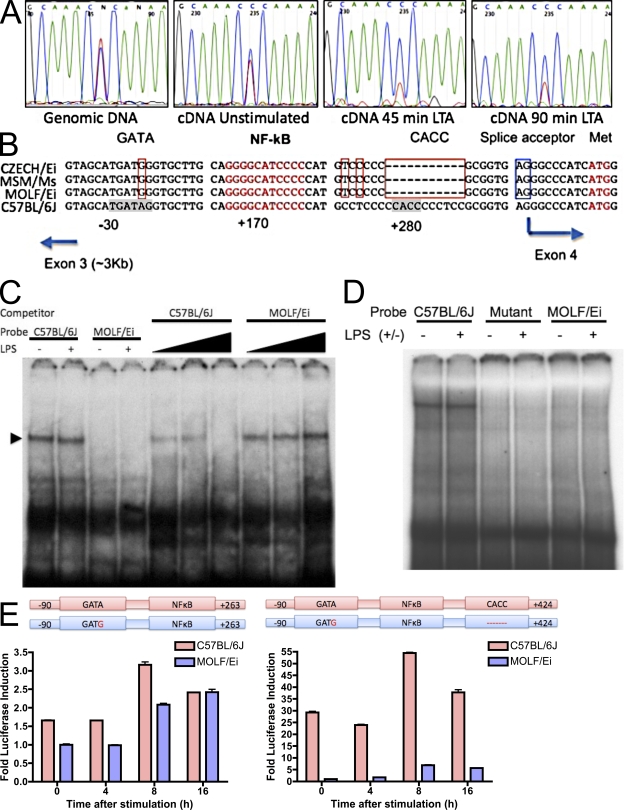
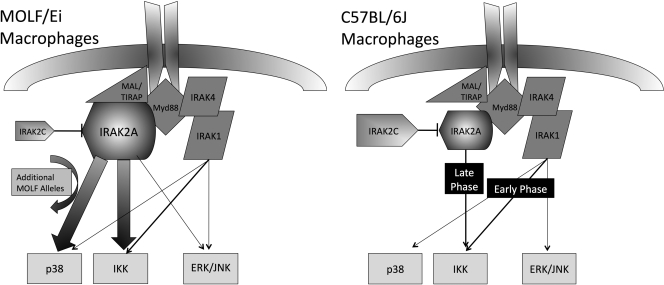
In a phenotypic screen of the wild-derived mouse strain MOLF/Ei, we describe an earlier and more potent toll-like receptor (TLR)-mediated induction of IL-6 transcription compared with the classical inbred strain C57BL/6J. The phenotype correlated with increased activity of the IkappaB kinase axis as well as p38, but not extracellular signal-regulated kinase or c-Jun N-terminal kinase, mitogen-activated protein kinase (MAPK) phosphorylation. The trait was mapped to the Why1 locus, which contains Irak2, a gene previously implicated as sustaining the late phase of TLR responses. In the MOLF/Ei TLR signaling network, IRAK-2 promotes early nuclear factor kappaB (NF-kappaB) activity and is essential for the activation of p38 MAPK. We identify a deletion in the MOLF/Ei promoter of the inhibitory Irak2c gene, leading to an increased ratio of pro- to antiinflammatory IRAK-2 isoforms. These findings demonstrate that IRAK-2 is an essential component of the early TLR response in MOLF/Ei mice and show a distinct pathway of p38 and NF-kappaB activation in this model organism. In addition, they demonstrate that studies in evolutionarily divergent model organisms are essential to complete dissection of signal transduction pathways.
Figures
References
-
- Akira S., Uematsu S., Takeuchi O. 2006. Pathogen recognition and innate immunity.Cell. 124:783–801 - PubMed
-
- Beebe A.M., Mauze S., Schork N.J., Coffman R.L. 1997. Serial backcross mapping of multiple loci associated with resistance to Leishmania major in mice.Immunity. 6:551–557 - PubMed
-
- Beutler B., Jiang Z., Georgel P., Crozat K., Croker B., Rutschmann S., Du X., Hoebe K. 2006. Genetic analysis of host resistance: toll-like receptor signaling and immunity at large.Annu. Rev. Immunol. 24:353–389 - PubMed
-
- Bonizzi G., Karin M. 2004. The two NF-kappaB activation pathways and their role in innate and adaptive immunity.Trends Immunol. 25:280–288 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
