

mGene: accurate SVM-based gene finding with an application to nematode genomes
- PMID: 19564452
- PMCID: PMC2775605
- DOI: 10.1101/gr.090597.108
mGene: accurate SVM-based gene finding with an application to nematode genomes
Abstract
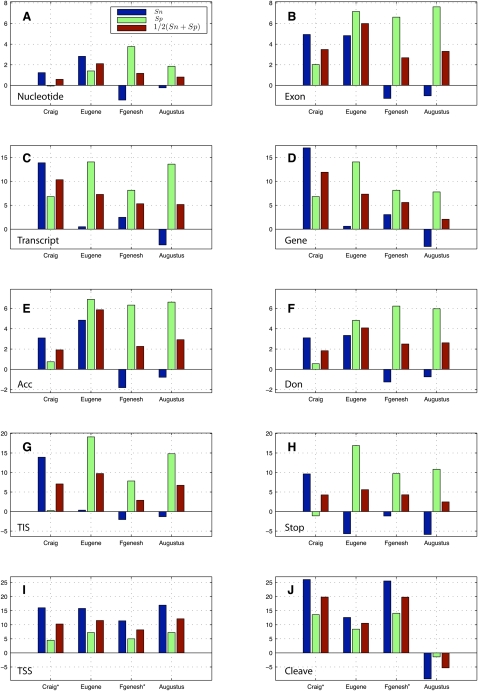
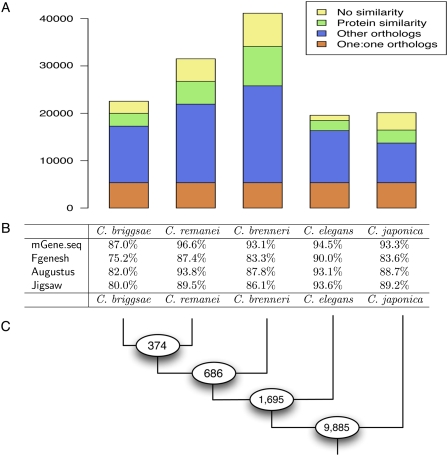
We present a highly accurate gene-prediction system for eukaryotic genomes, called mGene. It combines in an unprecedented manner the flexibility of generalized hidden Markov models (gHMMs) with the predictive power of modern machine learning methods, such as Support Vector Machines (SVMs). Its excellent performance was proved in an objective competition based on the genome of the nematode Caenorhabditis elegans. Considering the average of sensitivity and specificity, the developmental version of mGene exhibited the best prediction performance on nucleotide, exon, and transcript level for ab initio and multiple-genome gene-prediction tasks. The fully developed version shows superior performance in 10 out of 12 evaluation criteria compared with the other participating gene finders, including Fgenesh++ and Augustus. An in-depth analysis of mGene's genome-wide predictions revealed that approximately 2200 predicted genes were not contained in the current genome annotation. Testing a subset of 57 of these genes by RT-PCR and sequencing, we confirmed expression for 24 (42%) of them. mGene missed 300 annotated genes, out of which 205 were unconfirmed. RT-PCR testing of 24 of these genes resulted in a success rate of merely 8%. These findings suggest that even the gene catalog of a well-studied organism such as C. elegans can be substantially improved by mGene's predictions. We also provide gene predictions for the four nematodes C. briggsae, C. brenneri, C. japonica, and C. remanei. Comparing the resulting proteomes among these organisms and to the known protein universe, we identified many species-specific gene inventions. In a quality assessment of several available annotations for these genomes, we find that mGene's predictions are most accurate.
Figures

References
-
- Alexeyenko A, Tamas I, Liu G, Sonnhammer EL. Automatic clustering of orthologs and inparalogs shared by multiple proteomes. Bioinformatics. 2006;22:e9–e15. - PubMed
-
- Altun Y, Tsochantaridis I, Hofmann T. Hidden Markov support vector machines. Scientific Commons; St. Gallen, Switzerland: 2003.
MeSH terms
Substances
LinkOut - more resources
Full Text Sources