

Mitochondria and energetic depression in cell pathophysiology
- PMID: 19564950
- PMCID: PMC2695278
- DOI: 10.3390/ijms10052252
Mitochondria and energetic depression in cell pathophysiology
Abstract
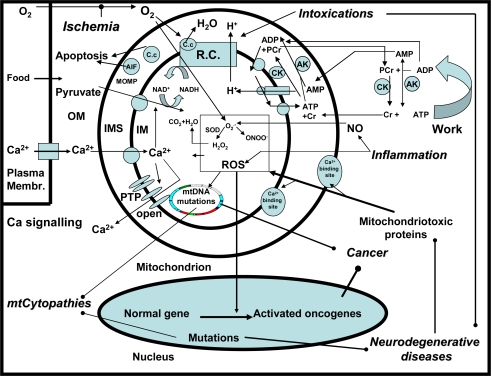
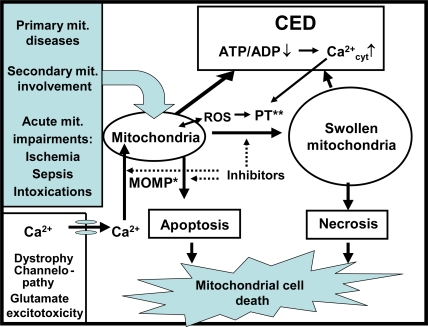
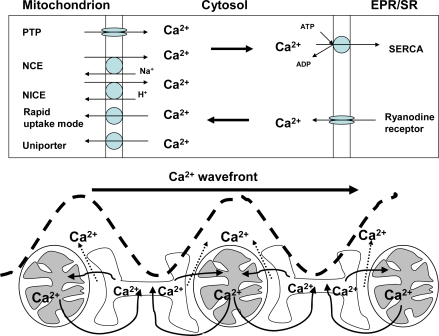
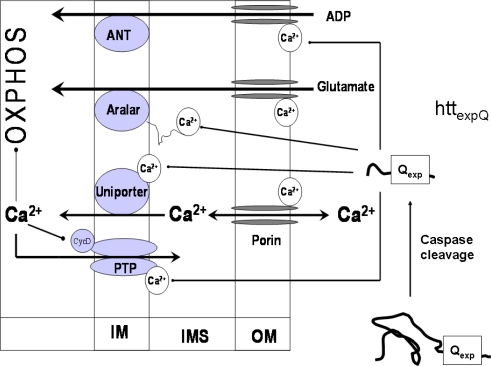
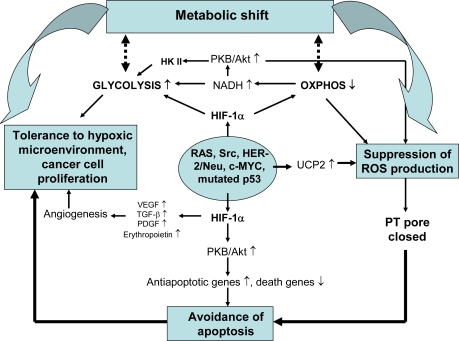
Mitochondrial dysfunction is a hallmark of almost all diseases. Acquired or inherited mutations of the mitochondrial genome DNA may give rise to mitochondrial diseases. Another class of disorders, in which mitochondrial impairments are initiated by extramitochondrial factors, includes neurodegenerative diseases and syndromes resulting from typical pathological processes, such as hypoxia/ischemia, inflammation, intoxications, and carcinogenesis. Both classes of diseases lead to cellular energetic depression (CED), which is characterized by decreased cytosolic phosphorylation potential that suppresses the cell's ability to do work and control the intracellular Ca(2+) homeostasis and its redox state. If progressing, CED leads to cell death, whose type is linked to the functional status of the mitochondria. In the case of limited deterioration, when some amounts of ATP can still be generated due to oxidative phosphorylation (OXPHOS), mitochondria launch the apoptotic cell death program by release of cytochrome c. Following pronounced CED, cytoplasmic ATP levels fall below the thresholds required for processing the ATP-dependent apoptotic cascade and the cell dies from necrosis. Both types of death can be grouped together as a mitochondrial cell death (MCD). However, there exist multiple adaptive reactions aimed at protecting cells against CED. In this context, a metabolic shift characterized by suppression of OXPHOS combined with activation of aerobic glycolysis as the main pathway for ATP synthesis (Warburg effect) is of central importance. Whereas this type of adaptation is sufficiently effective to avoid CED and to control the cellular redox state, thereby ensuring the cell survival, it also favors the avoidance of apoptotic cell death. This scenario may underlie uncontrolled cellular proliferation and growth, eventually resulting in carcinogenesis.
Keywords: cancer; energy depression; hypoxia; inflammation; mitochondria; mitochondrial cell death; neurodegenerative diseases.
Figures

References
-
- Warburg O, Geissler AW, Lorenz S. Genesis of tumor metabolism by vitamin B1 deficiency (thiamine deficiency) Z. Naturforsch. B. 1970;25:332–333. - PubMed
-
- Luft R. Luft's disease revisited. Severe hypermetabolism of nonthyroid origin with a defect in the maintenance of mitochondrial respiratory control. Mt. Sinai J. Med. 1992;59:140–145. - PubMed
-
- Holt IJ, Harding AE, Morgan-Hughes JA. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature. 1988;331:717–719. - PubMed
-
- Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM, Elsas LJ, Nikoskelainen EK. Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy. Science. 1988;242:1427–1430. - PubMed
-
- Triepels RH, Van Den Heuvel LP, Trijbels JM, Smeitink JA. Respiratory chain complex I deficiency. Am. J. Med. Gen. 2001;106:37–45. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
