

Death by committee: organellar trafficking and communication in apoptosis
- PMID: 19566895
- PMCID: PMC2746861
- DOI: 10.1111/j.1600-0854.2009.00951.x
Death by committee: organellar trafficking and communication in apoptosis
Abstract
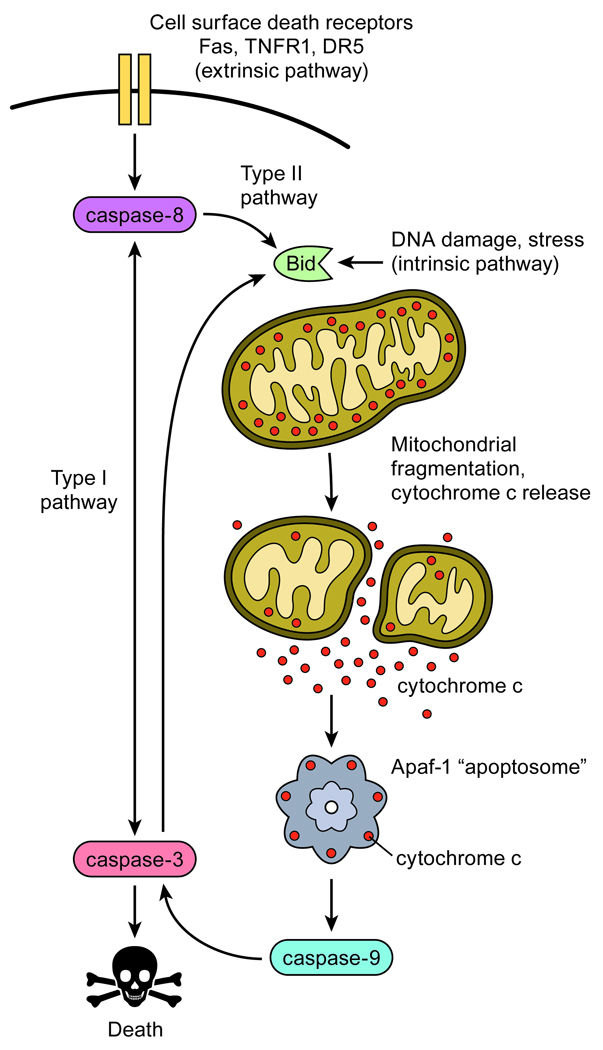
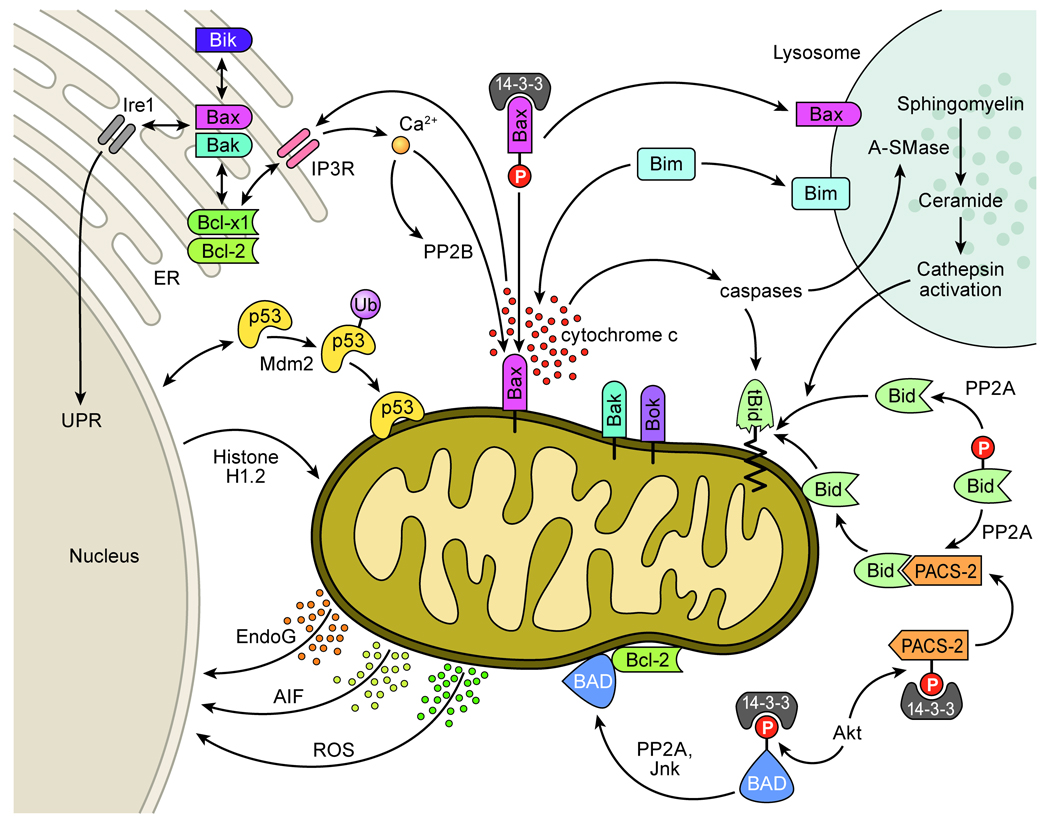
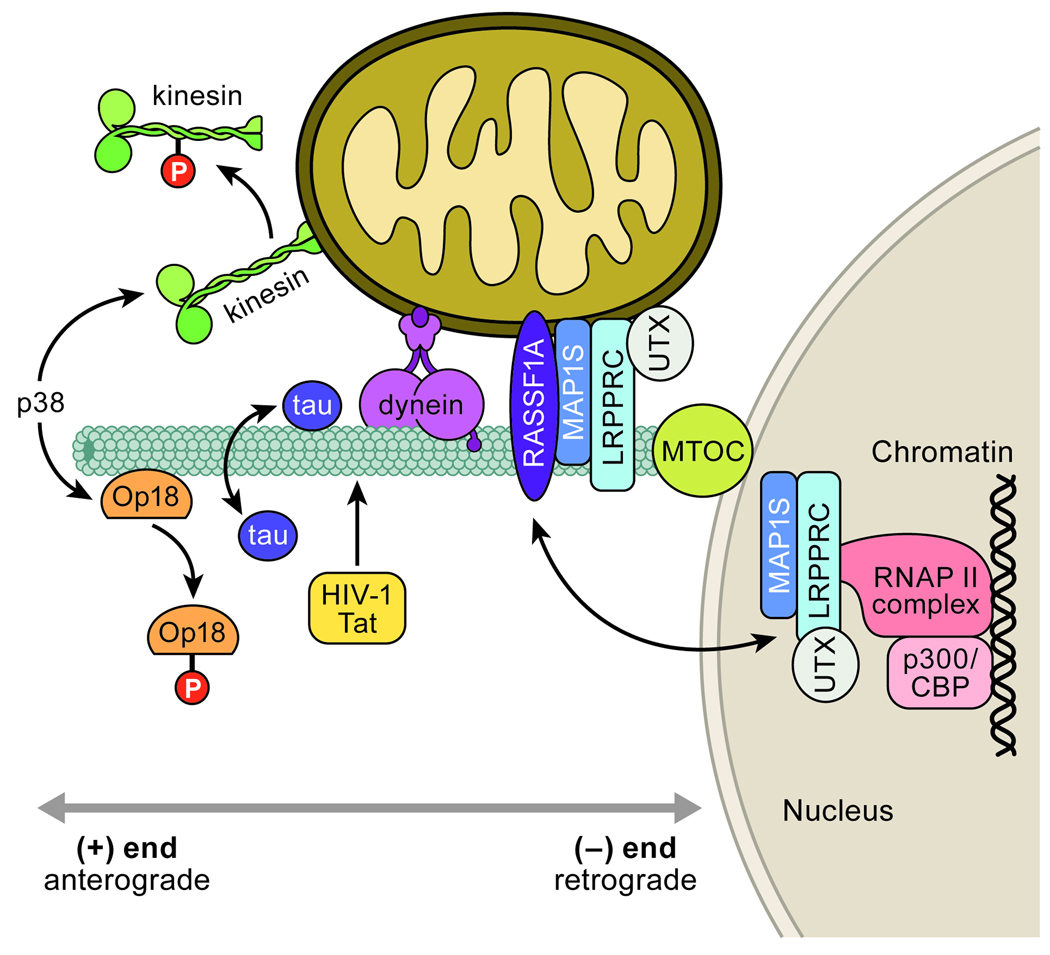
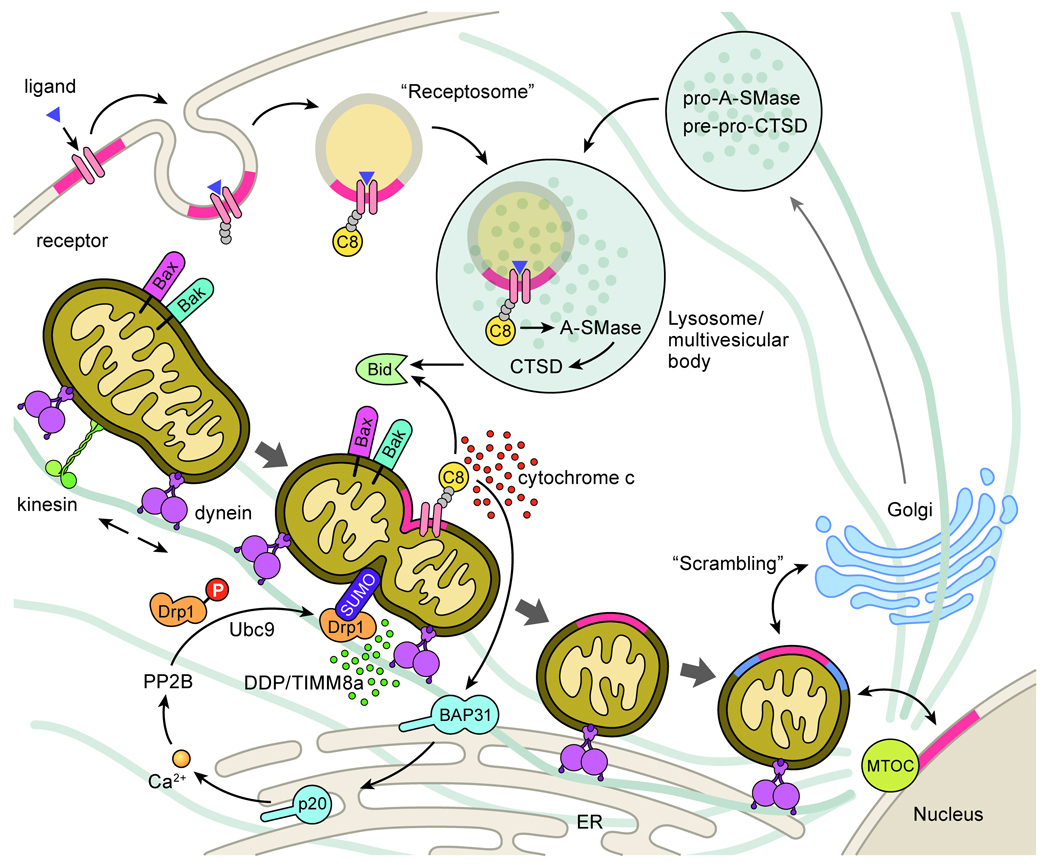
Apoptosis proceeds through a set of evolutionarily conserved processes that co-ordinate the elimination of damaged or unneeded cells. This program of cell death is carried out by organelle-directed regulators, including the Bcl-2 proteins, and ultimately executed by proteases of the caspase family. Although the biochemical mechanisms of apoptosis are increasingly understood, the underlying cell biology orchestrating programmed cell death remains enigmatic. In this review, we summarize the current understanding of Bcl-2 protein regulation and caspase activation while examining cell biological mechanisms and consequences of apoptotic induction. Organellar contributions to apoptotic induction include death receptor endocytosis, mitochondrial and lysosomal permeabilization, endoplasmic reticulum calcium release and fragmentation of the Golgi apparatus. These early apoptotic events are accompanied by stabilization of the microtubule cytoskeleton and translocation of organelles to the microtubule organizing center. Together, these phenomena establish a model of apoptotic induction whereby a cytoskeletal-dependent coalescence and 'scrambling' of organelles in the paranuclear region co-ordinates apoptotic communication, caspase activation and cell death.
Figures
References
-
- Degterev A, Yuan J. Expansion and evolution of cell death programmes. Nat Rev Mol Cell Biol. 2008 - PubMed
-
- Eckhart L, Ballaun C, Hermann M, Vandeberg JL, Sipos W, Uthman A, Fischer H, Tschachler E. Identification of novel mammalian caspases reveals an important role of gene loss in shaping the human caspase repertoire. Mol Biol Evol. 2008 - PubMed
-
- Luthi AU, Martin SJ. The CASBAH: a searchable database of caspase substrates. Cell Death Differ. 2007;14(4):641–650. - PubMed
-
- Peter ME, Krammer PH. The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ. 2003;10(1):26–35. - PubMed
-
- Ferri KF, Kroemer G. Organelle-specific initiation of cell death pathways. Nat Cell Biol. 2001;3(11):E255–E263. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
