

Extensions of sparse canonical correlation analysis with applications to genomic data
- PMID: 19572827
- PMCID: PMC2861323
- DOI: 10.2202/1544-6115.1470
Extensions of sparse canonical correlation analysis with applications to genomic data
Abstract
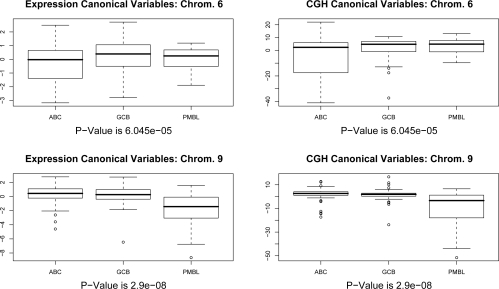
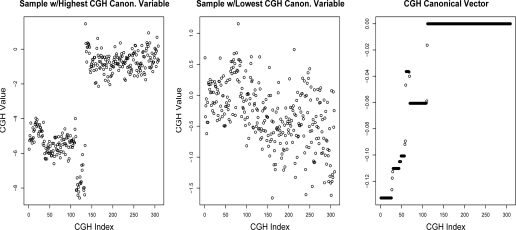
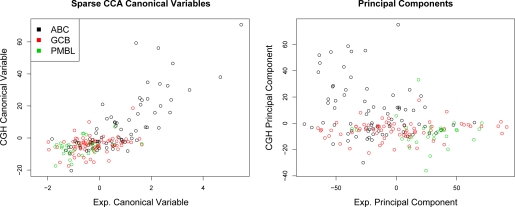
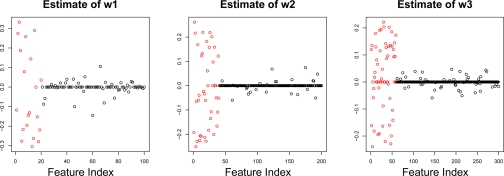
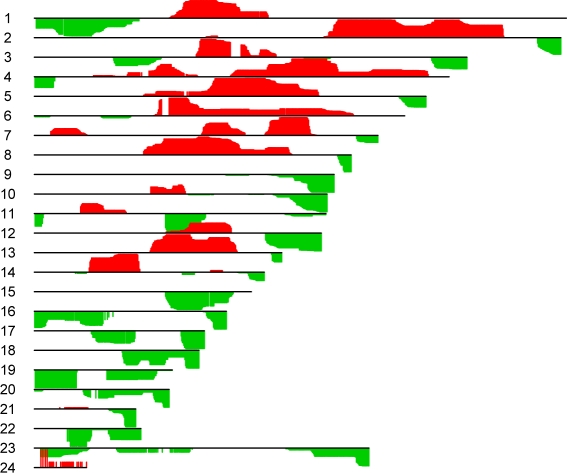
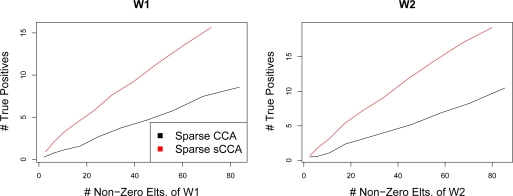
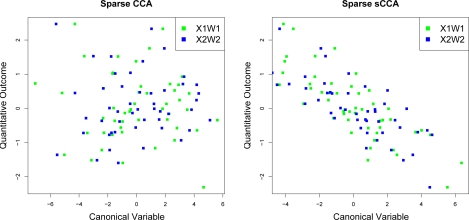
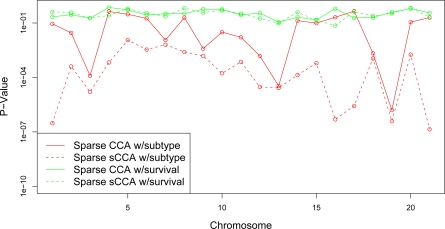
In recent work, several authors have introduced methods for sparse canonical correlation analysis (sparse CCA). Suppose that two sets of measurements are available on the same set of observations. Sparse CCA is a method for identifying sparse linear combinations of the two sets of variables that are highly correlated with each other. It has been shown to be useful in the analysis of high-dimensional genomic data, when two sets of assays are available on the same set of samples. In this paper, we propose two extensions to the sparse CCA methodology. (1) Sparse CCA is an unsupervised method; that is, it does not make use of outcome measurements that may be available for each observation (e.g., survival time or cancer subtype). We propose an extension to sparse CCA, which we call sparse supervised CCA, which results in the identification of linear combinations of the two sets of variables that are correlated with each other and associated with the outcome. (2) It is becoming increasingly common for researchers to collect data on more than two assays on the same set of samples; for instance, SNP, gene expression, and DNA copy number measurements may all be available. We develop sparse multiple CCA in order to extend the sparse CCA methodology to the case of more than two data sets. We demonstrate these new methods on simulated data and on a recently published and publicly available diffuse large B-cell lymphoma data set.
Figures
References
-
- Alizadeh A, Eisen M, Davis RE, Ma C, Lossos I, Rosenwald A, Boldrick J, Sabet H, Tran T, Yu X, Powell J, Marti G, Moore T, Hudson J, Lu L, Lewis D, Tibshirani R, Sherlock G, Chan W, Greiner T, Weisenburger D, Armitage K, Warnke R, Levy R, Wilson W, Grever M, Byrd J, Botstein D, Brown P, Staudt L. ‘Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling’. Nature. 2000;403:503–511. doi: 10.1038/35000501. - DOI - PubMed
-
- Bair E, Hastie T, Paul D, Tibshirani R. ‘Prediction by supervised principal components’. J Amer Statist Assoc. 2006;101:119–137. doi: 10.1198/016214505000000628. - DOI
-
- Gifi A. Nonlinear multivariate analysis. Wiley, Chichester; England: 1990.
-
- Hotelling H. ‘Relations between two sets of variates’. Biometrika. 1936;28:321–377.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous