

Deficiency of Dol-P-Man synthase subunit DPM3 bridges the congenital disorders of glycosylation with the dystroglycanopathies
- PMID: 19576565
- PMCID: PMC2706967
- DOI: 10.1016/j.ajhg.2009.06.006
Deficiency of Dol-P-Man synthase subunit DPM3 bridges the congenital disorders of glycosylation with the dystroglycanopathies
Abstract
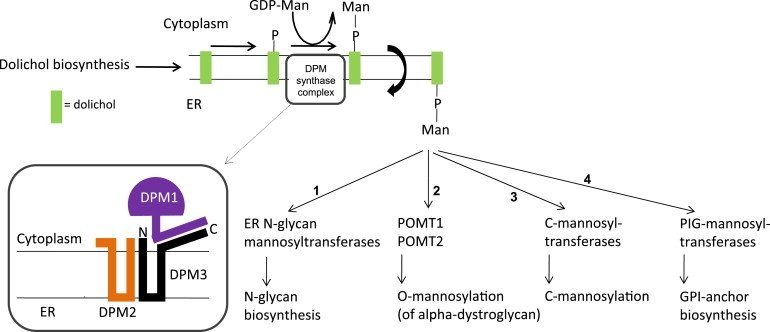
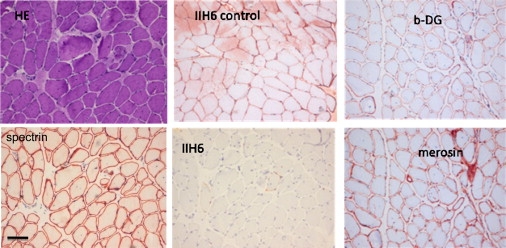
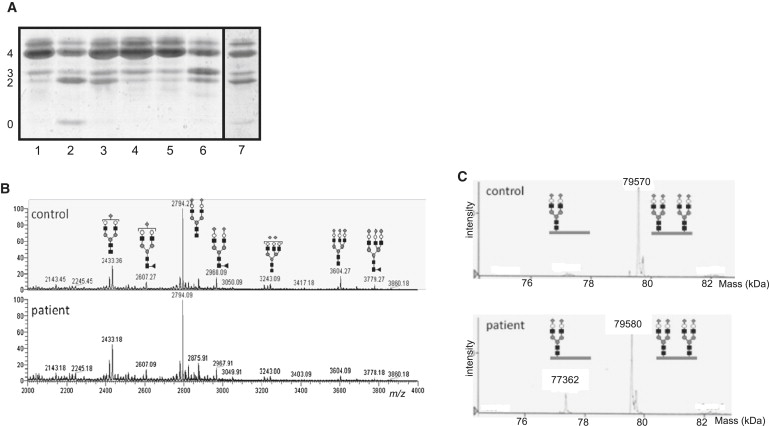
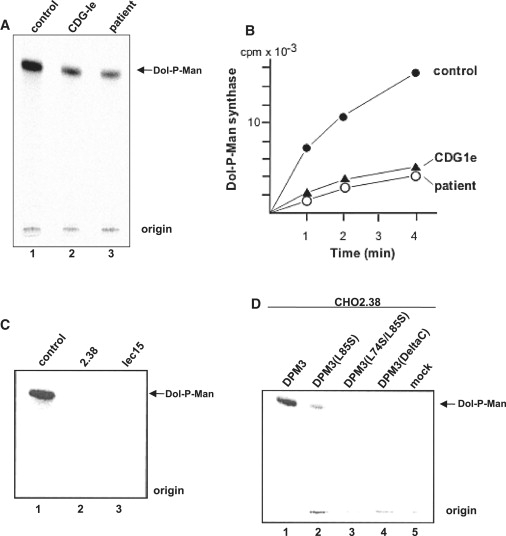
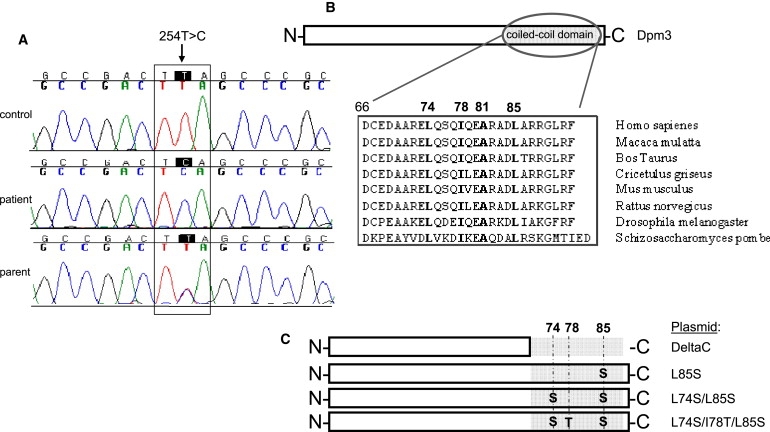
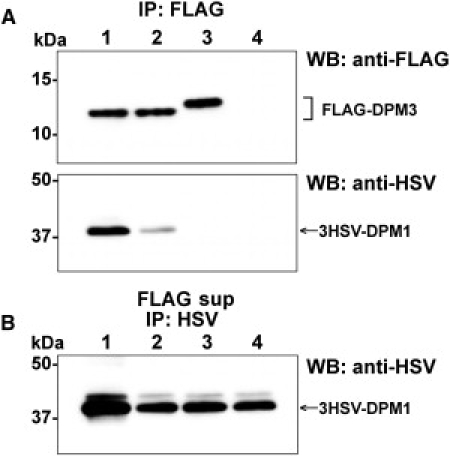
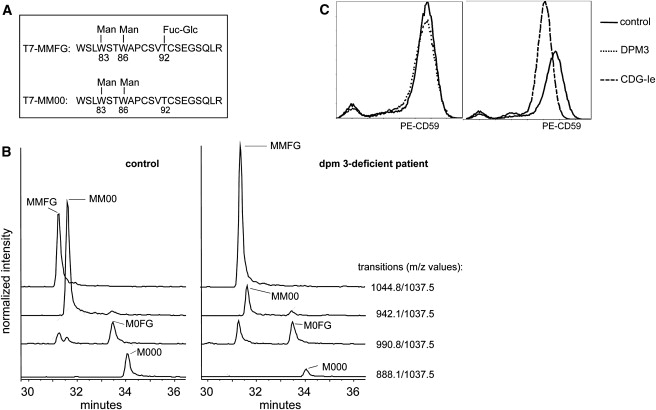
Alpha-dystroglycanopathies such as Walker Warburg syndrome represent an important subgroup of the muscular dystrophies that have been related to defective O-mannosylation of alpha-dystroglycan. In many patients, the underlying genetic etiology remains unsolved. Isolated muscular dystrophy has not been described in the congenital disorders of glycosylation (CDG) caused by N-linked protein glycosylation defects. Here, we present a genetic N-glycosylation disorder with muscular dystrophy in the group of CDG type I. Extensive biochemical investigations revealed a strongly reduced dolichol-phosphate-mannose (Dol-P-Man) synthase activity. Sequencing of the three DPM subunits and complementation of DPM3-deficient CHO2.38 cells showed a pathogenic p.L85S missense mutation in the strongly conserved coiled-coil domain of DPM3 that tethers catalytic DPM1 to the ER membrane. Cotransfection experiments in CHO cells showed a reduced binding capacity of DPM3(L85S) for DPM1. Investigation of the four Dol-P-Man-dependent glycosylation pathways in the ER revealed strongly reduced O-mannosylation of alpha-dystroglycan in a muscle biopsy, thereby explaining the clinical phenotype of muscular dystrophy. This mild Dol-P-Man biosynthesis defect due to DPM3 mutations is a cause for alpha-dystroglycanopathy, thereby bridging the congenital disorders of glycosylation with the dystroglycanopathies.
Figures
References
-
- Poppe M., Bourke J., Eagle M., Frosk P., Wrogemann K., Greenberg C., Muntoni F., Voit T., Straub V., Hilton-Jones D. Cardiac and respiratory failure in limb-girdle muscular dystrophy 2I. Ann. Neurol. 2004;56:738–741. - PubMed
-
- Brockington M., Yuva Y., Prandini P., Brown S.C., Torelli S., Benson M.A., Herrmann R., Anderson L.V., Bashir R., Burgunder J.M. Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum. Mol. Genet. 2001;10:2851–2859. - PubMed
-
- van Reeuwijk J., Brunner H.G., van Bokhoven H. Glyc-O-genetics of Walker-Warburg syndrome. Clin. Genet. 2005;67:281–289. - PubMed
-
- Mercuri E., Messina S., Bruno C., Mora M., Pegoraro E., Comi G.P., D'Amico A., Aiello C., Biancheri R., Berardinelli A. Congenital muscular dystrophies with defective glycosylation of dystroglycan. A population study. Neurology. 2009;72:1802–1809. - PubMed
-
- Godfrey C., Clement E., Mein R., Brockington M., Smith J., Talim B., Straub V., Robb S., Quinlivan R., Feng L. Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain. 2007;130:2725–2735. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
