

Cyclooxygenase-2 inhibition and hypoxia-induced pulmonary hypertension: effects on pulmonary vascular remodeling and contractility
- PMID: 19577709
- PMCID: PMC2746907
- DOI: 10.1016/j.tcm.2009.04.003
Cyclooxygenase-2 inhibition and hypoxia-induced pulmonary hypertension: effects on pulmonary vascular remodeling and contractility
Abstract
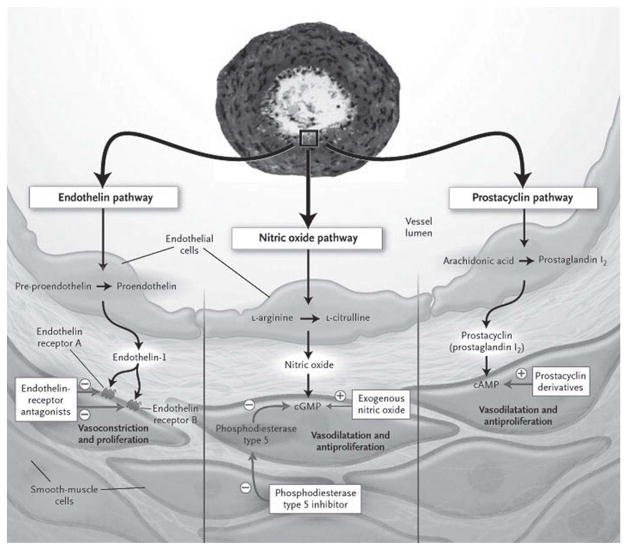
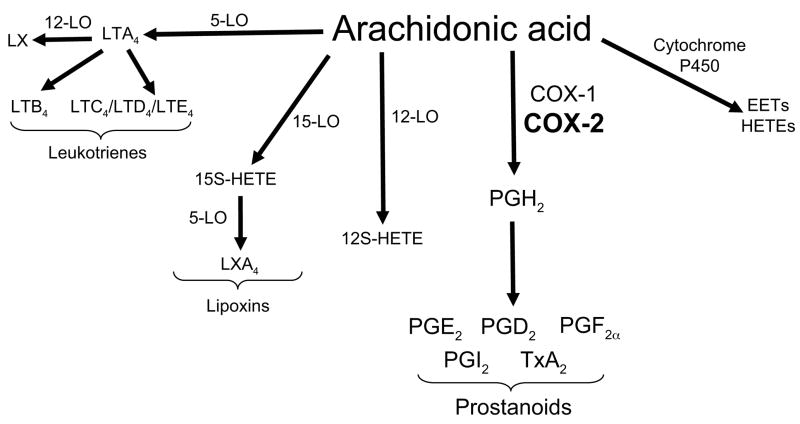
Pulmonary arterial hypertension (PAH) is a significant disease process characterized by elevated pulmonary vascular resistance leading to increased right ventricular afterload and ultimately progressing to right ventricular dysfunction and often death. Irreversible remodeling of the pulmonary vasculature is the hallmark of pulmonary hypertension and frequently leads to progressive functional decline in patients with PAH despite treatment with currently available therapies. Metabolites of the arachidonic acid cascade play an important homeostatic role in the pulmonary vasculature, and dysregulation of pathways downstream of arachidonic acid plays a central role in the pathobiology of PAH. Cyclooxygenase-2 (COX-2) is up-regulated in pulmonary artery smooth muscle cells (PASMC) and inflammatory cells during hypoxia and plays a protective role in the lung's response to hypoxia. We recently demonstrated that absence of COX-2 was detrimental in a mouse model of hypoxia-induced pulmonary hypertension. Exposure of COX-2 null mice to hypoxia resulted in severe pulmonary hypertension characterized by enhanced pulmonary vascular remodeling and significant up-regulation of the endothelin-1 receptor ET(A)R in the lung after hypoxia. Absence of COX-2 in vitro led to enhanced contractility of PASMC after exposure to hypoxia, which could be attenuated by iloprost, a prostaglandin I(2) analog. These findings suggest that selective inhibition of COX-2 may have detrimental pulmonary vascular consequences in patients with preexisting pulmonary hypertension or underlying hypoxemic lung diseases. Here, we discuss our recent data demonstrating the adverse consequences of COX-2 inhibition on pulmonary vascular remodeling and PASMC contractility.
Figures
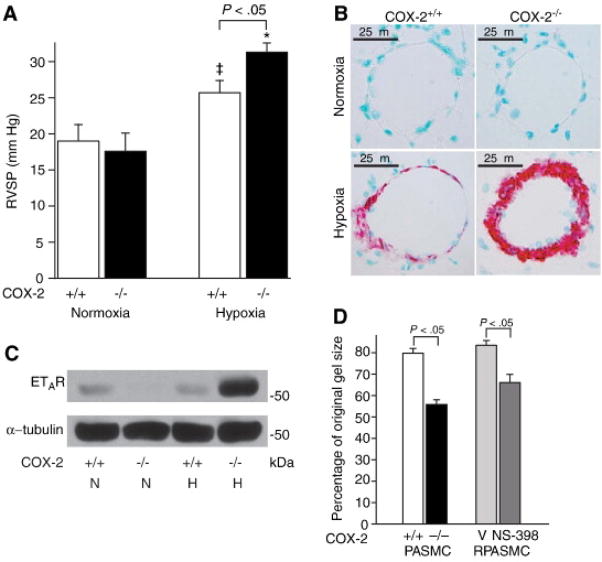
 and NS-398
and NS-398  ) exposed to hypoxia. Data are expressed as mean ± SE (p<0.05 for COX-2−/− PASMC vs. COX-2+/+ PASMC; p<0.05 for NS-398-treated vs. vehicle-treated RPASMC).
) exposed to hypoxia. Data are expressed as mean ± SE (p<0.05 for COX-2−/− PASMC vs. COX-2+/+ PASMC; p<0.05 for NS-398-treated vs. vehicle-treated RPASMC).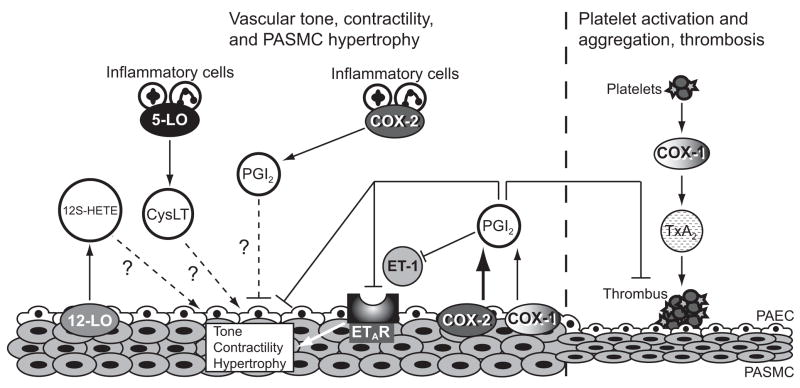
References
-
- Aldred MA, Vijayakrishnan J, James V, Soubrier F, Gomez-Sanchez MA, Martensson G, et al. BMPR2 gene rearrangements account for a significant proportion of mutations in familial and idiopathic pulmonary arterial hypertension. Hum Mutat. 2006;27:212–213. - PubMed
-
- Bos JL. Epac proteins: multi-purpose cAMP targets. Trends Biochem Sci. 2006;31:680–686. - PubMed
-
- Cathcart MC, Tamosiuniene R, Chen G, Neilan TG, Bradford A, O’Byrne KJ, et al. Cyclooxygenase-2-linked attenuation of hypoxia-induced pulmonary hypertension and intravascular thrombosis. J Pharmacol Exp Ther. 2008;326:51–58. - PubMed
-
- Chang HC, Weng CF. Cyclooxygenase-2 level and culture conditions influence NS398-induced apoptosis and caspase activation in lung cancer cells. Oncol Rep. 2001;8:1321–1325. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
