

BiGGEsTS: integrated environment for biclustering analysis of time series gene expression data
- PMID: 19583847
- PMCID: PMC2720980
- DOI: 10.1186/1756-0500-2-124
BiGGEsTS: integrated environment for biclustering analysis of time series gene expression data
Abstract
Background: The ability to monitor changes in expression patterns over time, and to observe the emergence of coherent temporal responses using expression time series, is critical to advance our understanding of complex biological processes. Biclustering has been recognized as an effective method for discovering local temporal expression patterns and unraveling potential regulatory mechanisms. The general biclustering problem is NP-hard. In the case of time series this problem is tractable, and efficient algorithms can be used. However, there is still a need for specialized applications able to take advantage of the temporal properties inherent to expression time series, both from a computational and a biological perspective.
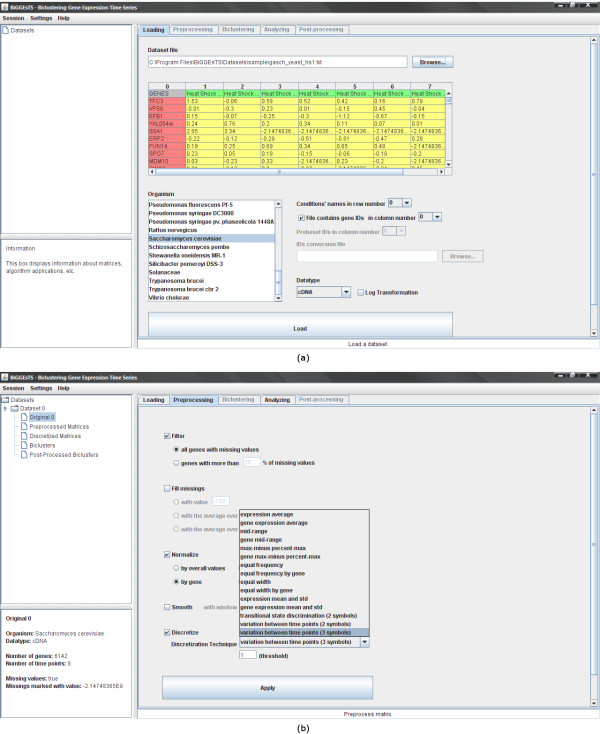
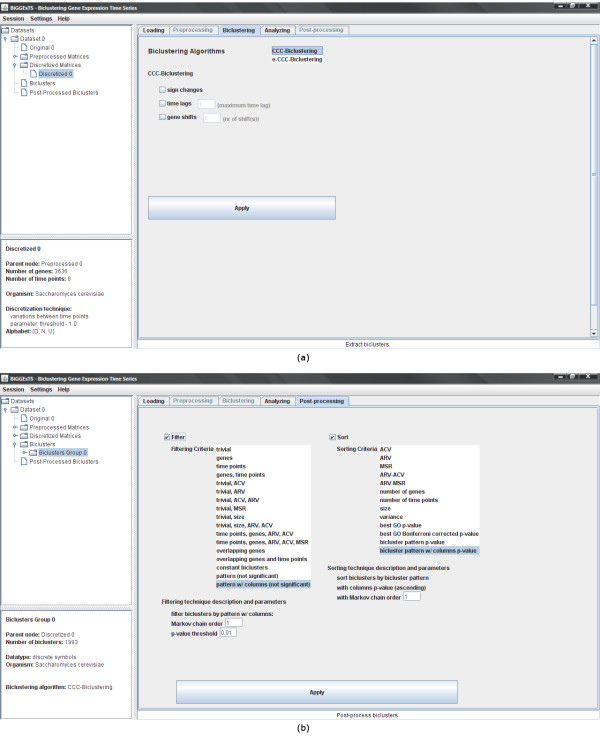
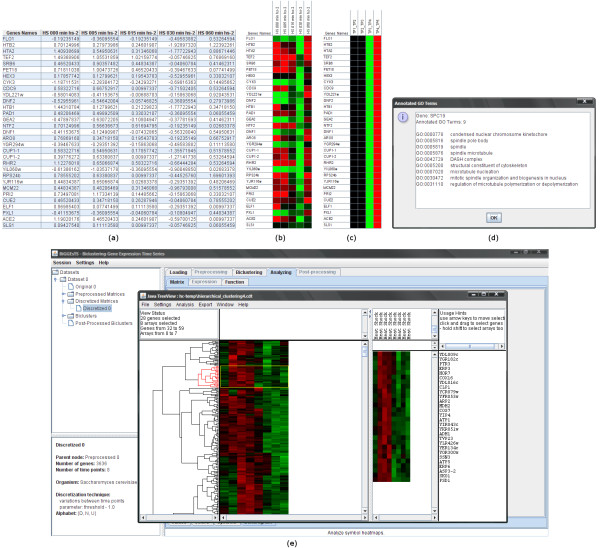
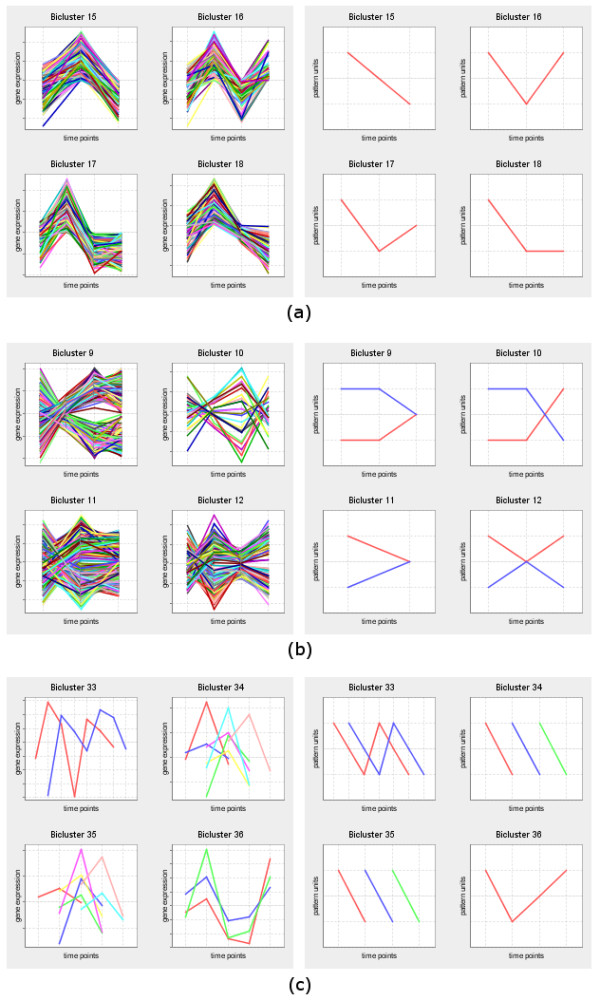
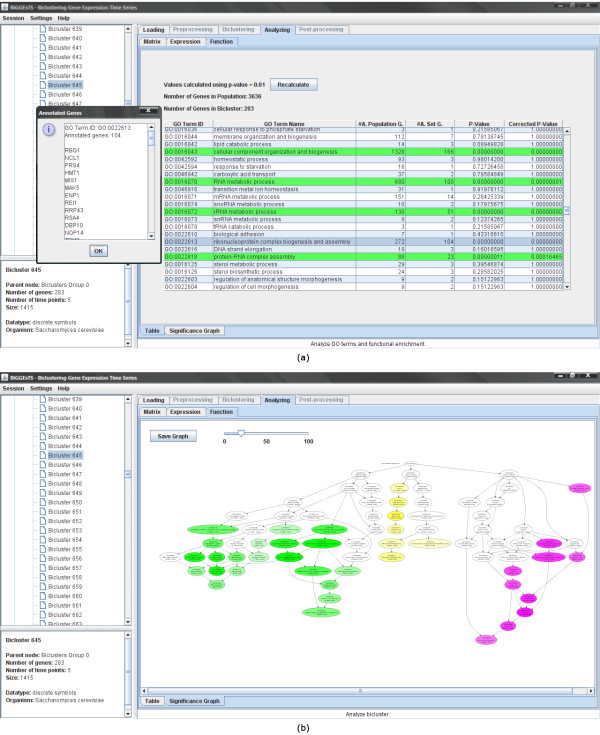
Findings: BiGGEsTS makes available state-of-the-art biclustering algorithms for analyzing expression time series. Gene Ontology (GO) annotations are used to assess the biological relevance of the biclusters. Methods for preprocessing expression time series and post-processing results are also included. The analysis is additionally supported by a visualization module capable of displaying informative representations of the data, including heatmaps, dendrograms, expression charts and graphs of enriched GO terms.
Conclusion: BiGGEsTS is a free open source graphical software tool for revealing local coexpression of genes in specific intervals of time, while integrating meaningful information on gene annotations. It is freely available at: http://kdbio.inesc-id.pt/software/biggests. We present a case study on the discovery of transcriptional regulatory modules in the response of Saccharomyces cerevisiae to heat stress.
Figures
References
-
- Cheng Y, Church GM. Biclustering of expression data. Proc Int Conf Intell Syst Mol Biol. 2000;8:93–103. - PubMed
-
- Ji L, Tan K. Identifying time-lagged gene clusters using gene expression data. Bioinformatics. 2005;21:509–516. - PubMed
-
- Zhang Y, Zha H, Chu CH. Proc of the 5th IEEE International Conference on Information Technology: Coding and Computing. Las Vegas, Nevada, USA: IEEE Computer Society; 2005. A time-series biclustering algorithm for revealing co-regulated genes; pp. 32–37.
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
