

Anionic pulmonary surfactant phospholipids inhibit inflammatory responses from alveolar macrophages and U937 cells by binding the lipopolysaccharide-interacting proteins CD14 and MD-2
- PMID: 19584052
- PMCID: PMC2757950
- DOI: 10.1074/jbc.M109.040832
Anionic pulmonary surfactant phospholipids inhibit inflammatory responses from alveolar macrophages and U937 cells by binding the lipopolysaccharide-interacting proteins CD14 and MD-2
Abstract
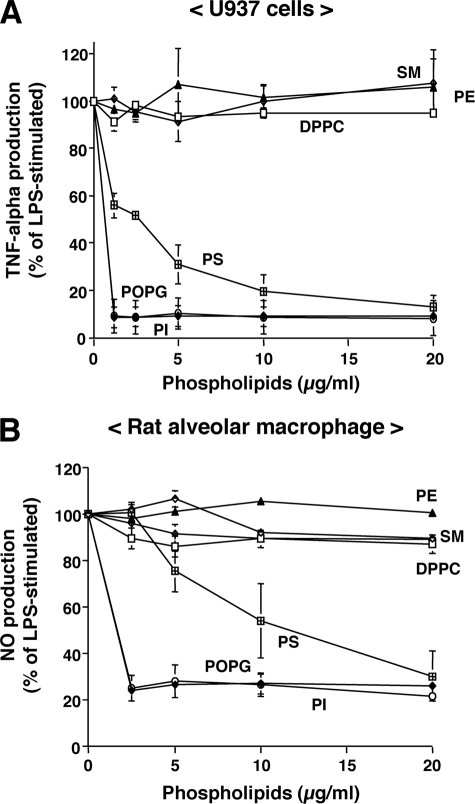
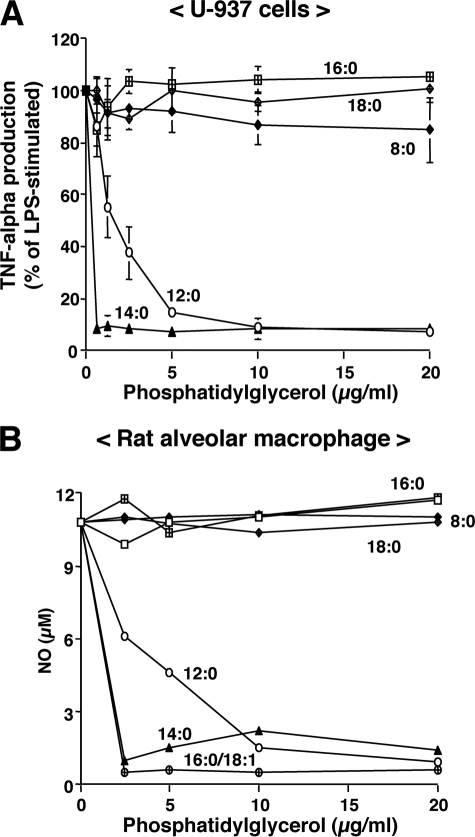
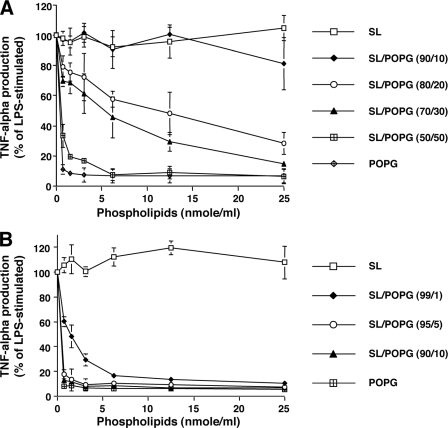
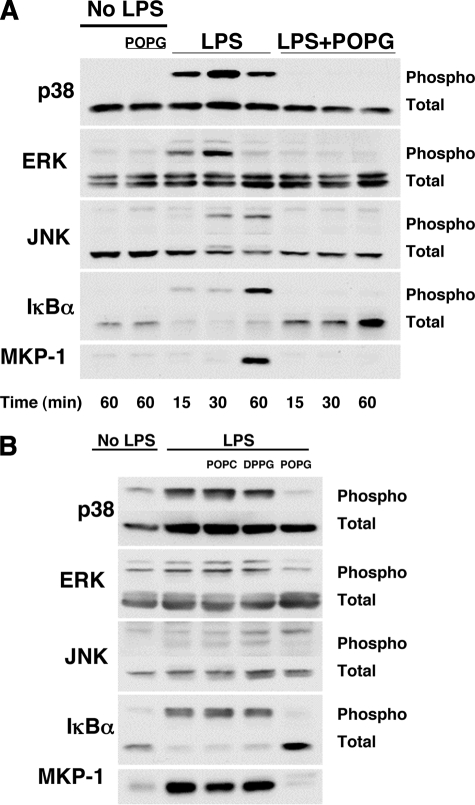
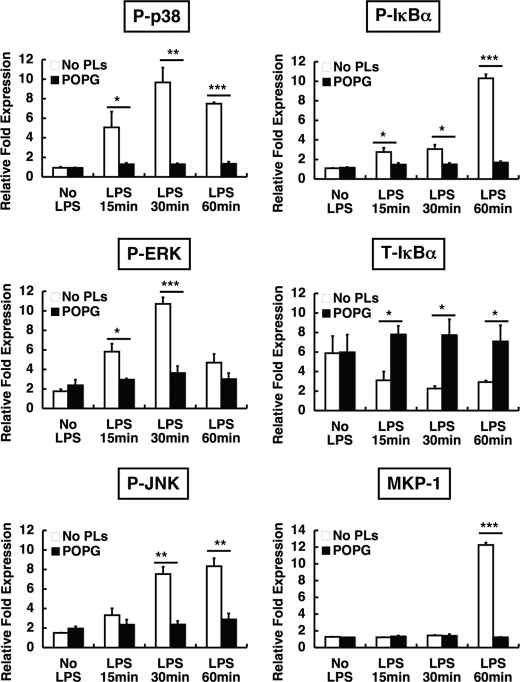
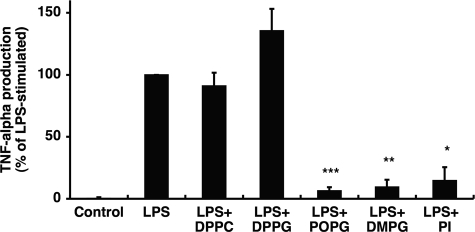
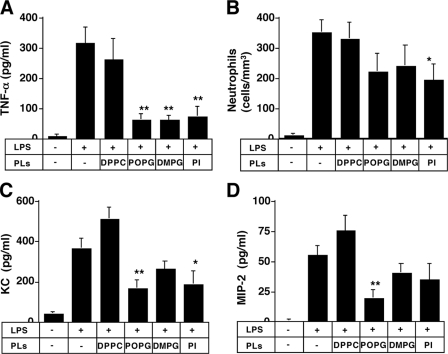
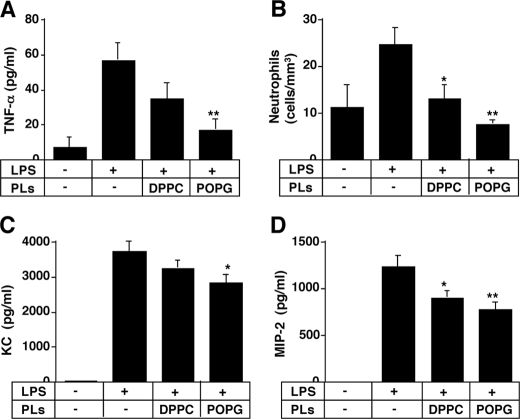
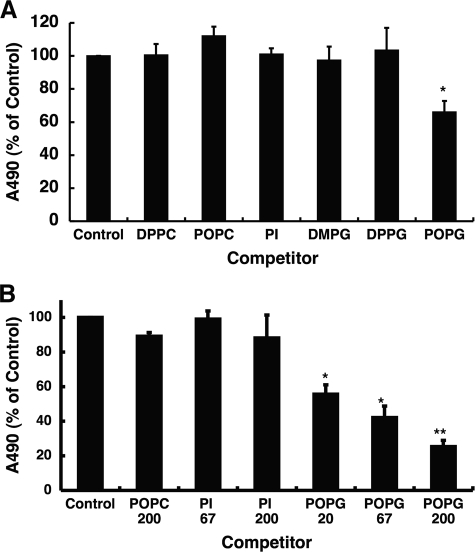
Lipopolysaccharide (LPS), derived from Gram-negative bacteria, is a major cause of acute lung injury and respiratory distress syndrome. Pulmonary surfactant is secreted as a complex mixture of lipids and proteins onto the alveolar surface of the lung. Surfactant phospholipids are essential in reducing surface tension at the air-liquid interface and preventing alveolar collapse at the end of the respiratory cycle. In the present study, we determined that palmitoyl-oleoyl-phosphatidylglycerol and phosphatidylinositol, which are minor components of pulmonary surfactant, and synthetic dimyristoylphosphatidylglycerol regulated the inflammatory response of alveolar macrophages. The anionic lipids significantly inhibited LPS-induced nitric oxide and tumor necrosis factor-alpha production from rat and human alveolar macrophages and a U937 cell line by reducing the LPS-elicited phosphorylation of multiple intracellular protein kinases. The anionic lipids were also effective at attenuating inflammation when administered intratracheally to mice challenged with LPS. Binding studies revealed high affinity interactions between the palmitoyl-oleoyl-phosphatidylglycerol and the Toll-like receptor 4-interacting proteins CD14 and MD-2. Our data clearly identify important anti-inflammatory properties of the minor surfactant phospholipids at the environmental interface of the lung.
Figures
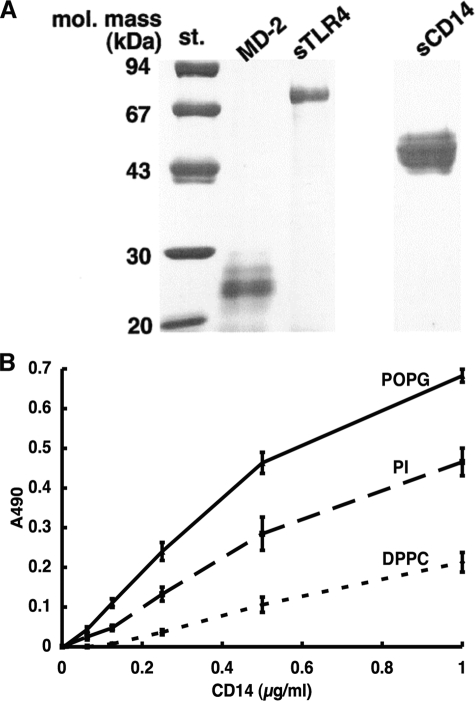
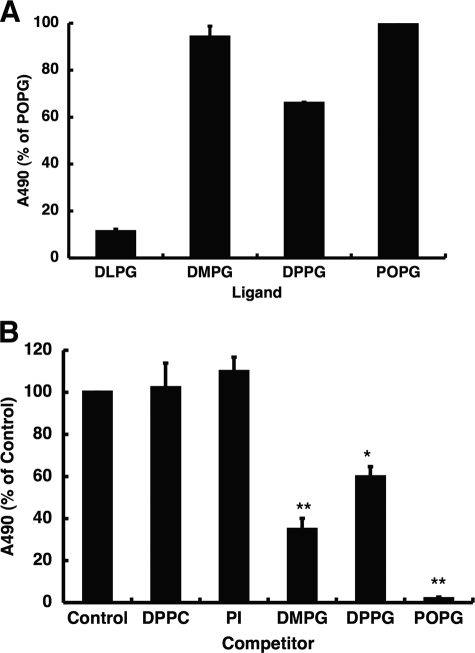
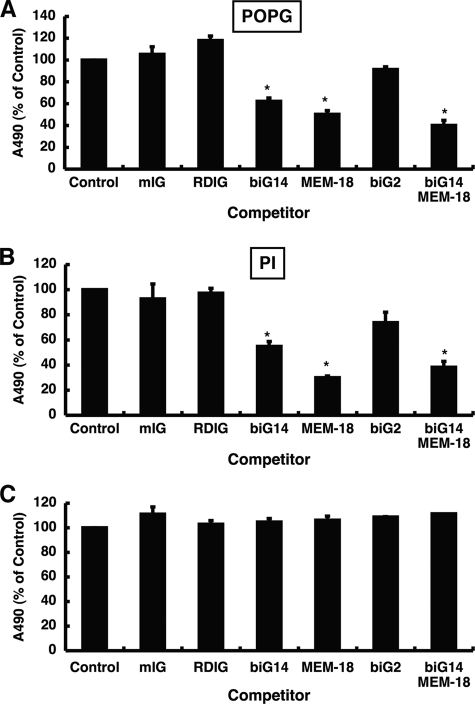
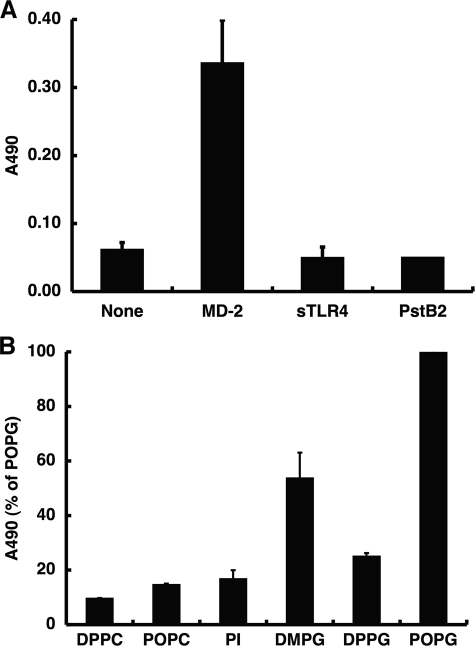
References
-
- O'Brien A. D., Rosenstreich D. L., Scher I., Campbell G. H., MacDermott R. P., Formal S. B. (1980) J. Immunol. 124, 20–24 - PubMed
-
- Ulevitch R. J., Tobias P. S. (1995) Annu. Rev. Immunol. 13, 437–457 - PubMed
-
- Wright S. D., Ramos R. A., Tobias P. S., Ulevitch R. J., Mathison J. C. (1990) Science 249, 1431–1433 - PubMed
-
- Nagai Y., Akashi S., Nagafuku M., Ogata M., Iwakura Y., Akira S., Kitamura T., Kosugi A., Kimoto M., Miyake K. (2002) Nat. Immunol. 3, 667–672 - PubMed
-
- Poltorak A., He X., Smirnova I., Liu M. Y., Van Huffel C., Du X., Birdwell D., Alejos E., Silva M., Galanos C., Freudenberg M., Ricciardi-Castagnoli P., Layton B., Beutler B. (1998) Science 282, 2085–2088 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
