

A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing's sarcoma
- PMID: 19584866
- PMCID: PMC2777681
- DOI: 10.1038/nm.1983
A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing's sarcoma
Abstract
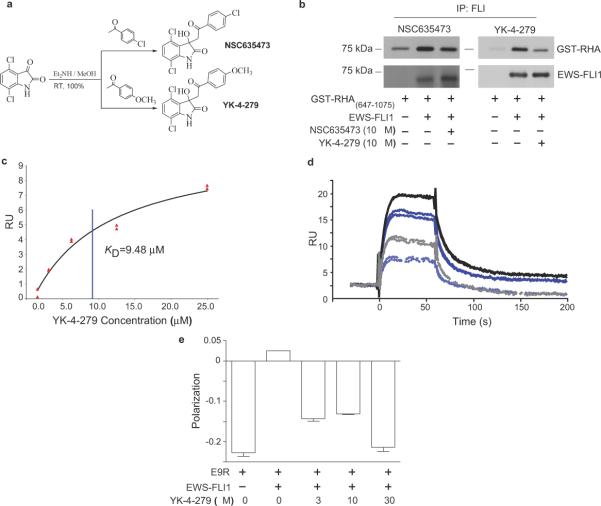
Many sarcomas and leukemias carry nonrandom chromosomal translocations encoding tumor-specific mutant fusion transcription factors that are essential to their molecular pathogenesis. Ewing's sarcoma family tumors (ESFTs) contain a characteristic t(11;22) translocation leading to expression of the oncogenic fusion protein EWS-FLI1. EWS-FLI1 is a disordered protein that precludes standard structure-based small-molecule inhibitor design. EWS-FLI1 binding to RNA helicase A (RHA) is important for its oncogenic function. We therefore used surface plasmon resonance screening to identify compounds that bind EWS-FLI1 and might block its interaction with RHA. YK-4-279, a derivative of the lead compound from the screen, blocks RHA binding to EWS-FLI1, induces apoptosis in ESFT cells and reduces the growth of ESFT orthotopic xenografts. These findings provide proof of principle that inhibiting the interaction of mutant cancer-specific transcription factors with the normal cellular binding partners required for their oncogenic activity provides a promising strategy for the development of uniquely effective, tumor-specific anticancer agents.
Figures





References
-
- Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer. 2007;7:233–245. - PubMed
-
- French CA, et al. Midline carcinoma of children and young adults with NUT rearrangement. J Clin Oncol. 2004;22:4135–4139. - PubMed
-
- Helman LJ, Meltzer P. Mechanisms of sarcoma development. Nat Rev Cancer. 2003;3:685–694. - PubMed
-
- Poppe B, et al. Expression analyses identify MLL as a prominent target of 11q23 amplification and support an etiologic role for MLL gain of function in myeloid malignancies. Blood. 2004;103:229–235. - PubMed
-
- Carroll M, et al. CGP 57148, a tyrosine kinase inhibitor, inhibits the growth of cells expressing BCR-ABL, TEL-ABL, and TEL-PDGFR fusion proteins. Blood. 1997;90:4947–4952. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
