

HaMStR: profile hidden markov model based search for orthologs in ESTs
- PMID: 19586527
- PMCID: PMC2723089
- DOI: 10.1186/1471-2148-9-157
HaMStR: profile hidden markov model based search for orthologs in ESTs
Abstract
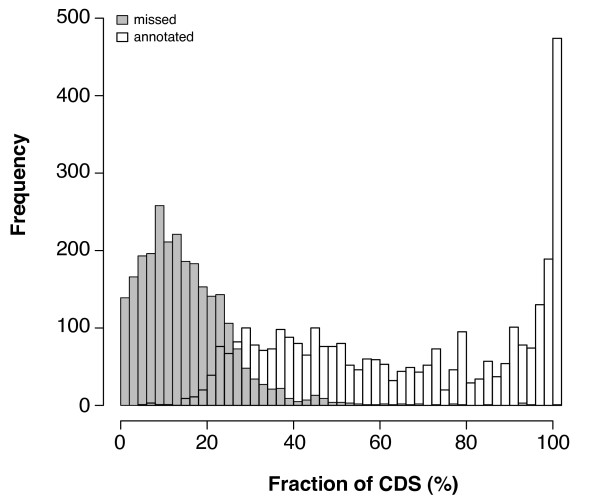
Background: EST sequencing is a versatile approach for rapidly gathering protein coding sequences. They provide direct access to an organism's gene repertoire bypassing the still error-prone procedure of gene prediction from genomic data. Therefore, ESTs are often the only source for biological sequence data from taxa outside mainstream interest. The widespread use of ESTs in evolutionary studies and particularly in molecular systematics studies is still hindered by the lack of efficient and reliable approaches for automated ortholog predictions in ESTs. Existing methods either depend on a known species tree or cannot cope with redundancy in EST data.
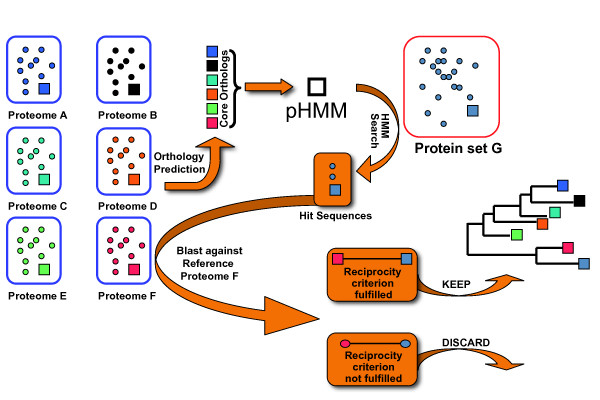
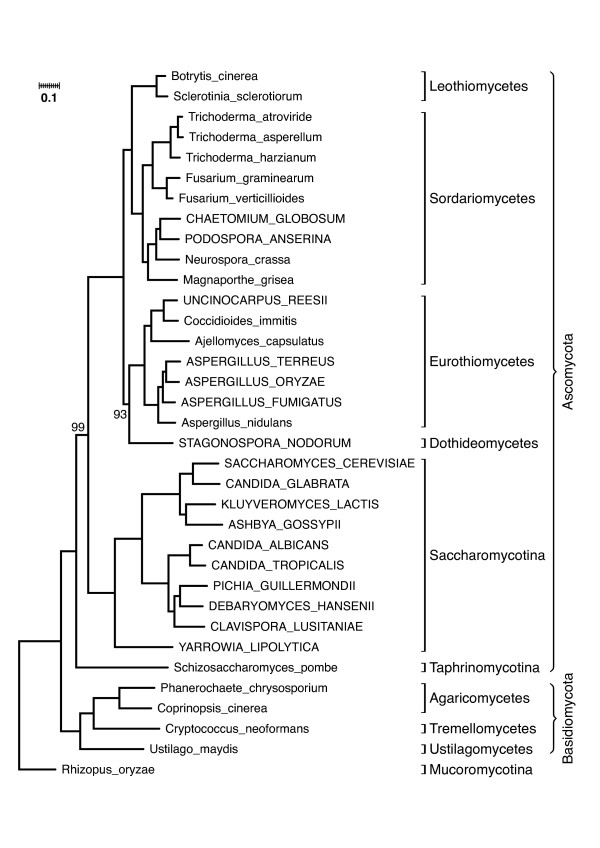
Results: We present a novel approach (HaMStR) to mine EST data for the presence of orthologs to a curated set of genes. HaMStR combines a profile Hidden Markov Model search and a subsequent BLAST search to extend existing ortholog cluster with sequences from further taxa. We show that the HaMStR results are consistent with those obtained with existing orthology prediction methods that require completely sequenced genomes. A case study on the phylogeny of 35 fungal taxa illustrates that HaMStR is well suited to compile informative data sets for phylogenomic studies from ESTs and protein sequence data.
Conclusion: HaMStR extends in a standardized manner a pre-defined set of orthologs with ESTs from further taxa. In the same fashion HaMStR can be applied to protein sequence data, and thus provides a comprehensive approach to compile ortholog cluster from any protein coding data. The resulting orthology predictions serve as the data basis for a variety of evolutionary studies. Here, we have demonstrated the application of HaMStR in a molecular systematics study. However, we envision that studies tracing the evolutionary fate of individual genes or functional complexes of genes will greatly benefit from HaMStR orthology predictions as well.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
