

Lysophosphatidic acid enhances pulmonary epithelial barrier integrity and protects endotoxin-induced epithelial barrier disruption and lung injury
- PMID: 19586906
- PMCID: PMC2782006
- DOI: 10.1074/jbc.M109.007393
Lysophosphatidic acid enhances pulmonary epithelial barrier integrity and protects endotoxin-induced epithelial barrier disruption and lung injury
Abstract
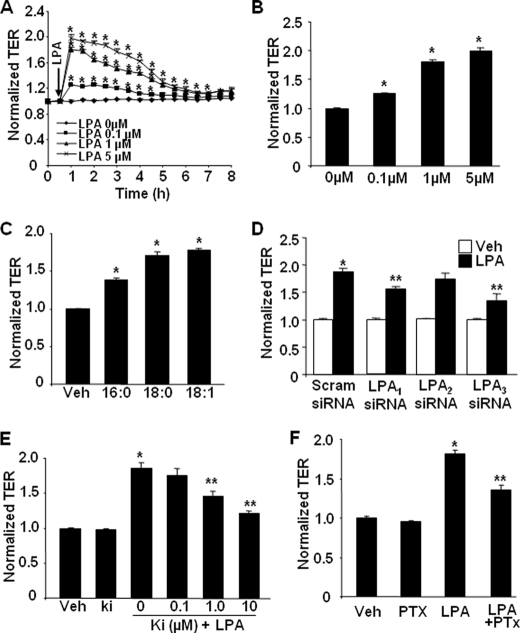
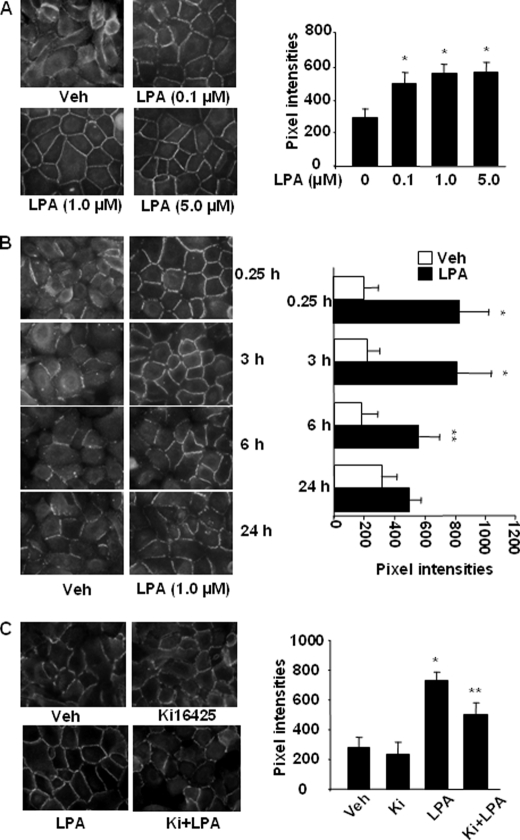
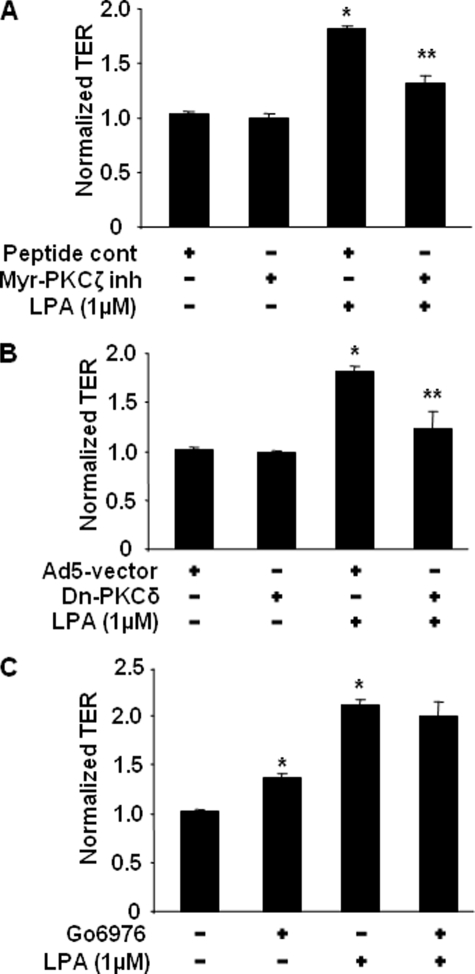
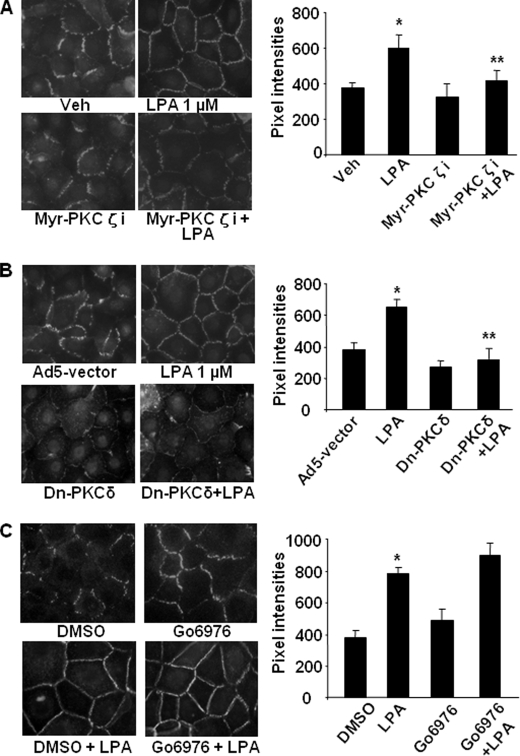
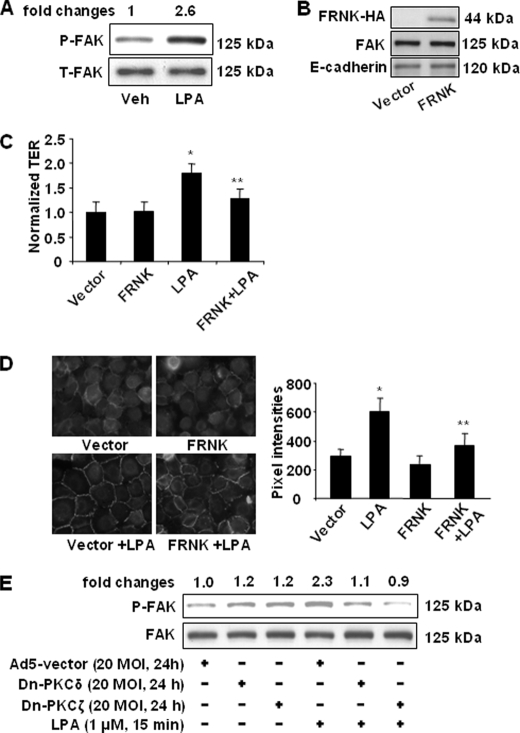
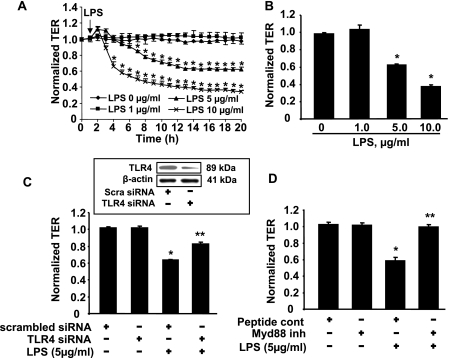
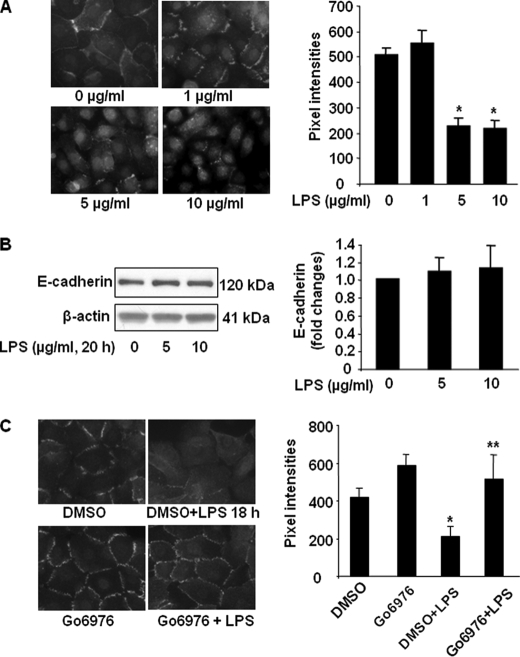
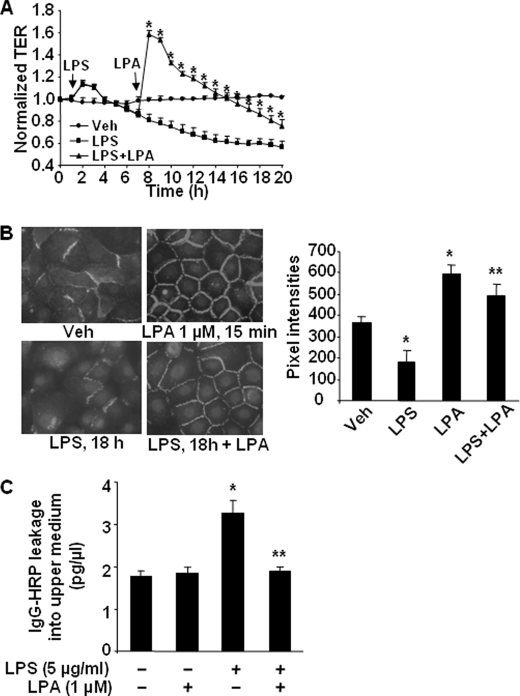
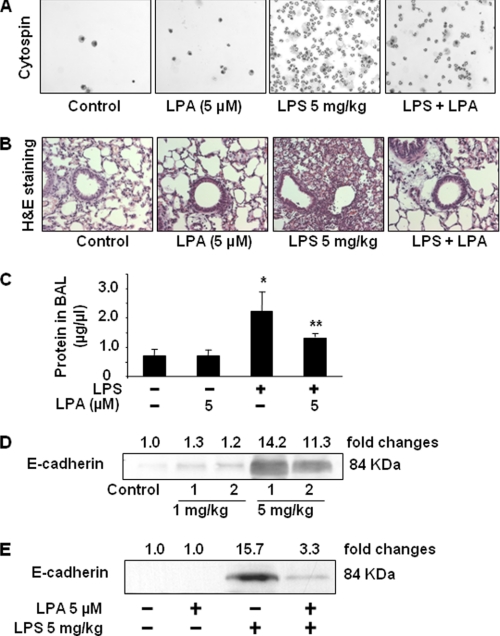
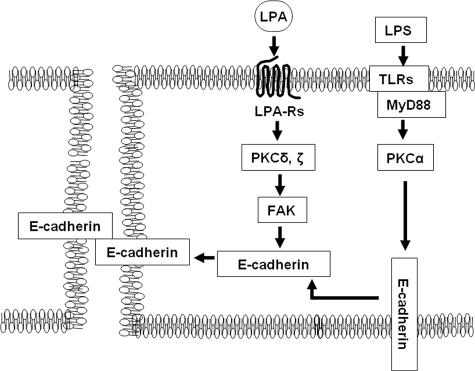
Lysophosphatidic acid (LPA), a bioactive phospholipid, induces a wide range of cellular effects, including gene expression, cytoskeletal rearrangement, and cell survival. We have previously shown that LPA stimulates secretion of pro- and anti-inflammatory cytokines in bronchial epithelial cells. This study provides evidence that LPA enhances pulmonary epithelial barrier integrity through protein kinase C (PKC) delta- and zeta-mediated E-cadherin accumulation at cell-cell junctions. Treatment of human bronchial epithelial cells (HBEpCs) with LPA increased transepithelial electrical resistance (TER) by approximately 2.0-fold and enhanced accumulation of E-cadherin to the cell-cell junctions through Galpha(i)-coupled LPA receptors. Knockdown of E-cadherin with E-cadherin small interfering RNA or pretreatment with EGTA (0.1 mm) prior to LPA (1 microm) treatment attenuated LPA-induced increases in TER in HBEpCs. Furthermore, LPA induced tyrosine phosphorylation of focal adhesion kinase (FAK) and overexpression of the FAK inhibitor, and FAK-related non-kinase-attenuated LPA induced increases in TER and E-cadherin accumulation at cell-cell junctions. Overexpression of dominant negative protein kinase delta and zeta attenuated LPA-induced phosphorylation of FAK, accumulation of E-cadherin at cell-cell junctions, and an increase in TER. Additionally, lipopolysaccharide decreased TER and induced E-cadherin relocalization from cell-cell junctions to cytoplasm in a dose-dependent fashion, which was restored by LPA post-treatment in HBEpCs. Intratracheal post-treatment with LPA (5 microm) reduced LPS-induced neutrophil influx, protein leak, and E-cadherin shedding in bronchoalveolar lavage fluids in a murine model of acute lung injury. These data suggest a protective role of LPA in airway inflammation and remodeling.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
