

Disruption of LANA in rhesus rhadinovirus generates a highly lytic recombinant virus
- PMID: 19587030
- PMCID: PMC2748027
- DOI: 10.1128/JVI.00704-09
Disruption of LANA in rhesus rhadinovirus generates a highly lytic recombinant virus
Abstract
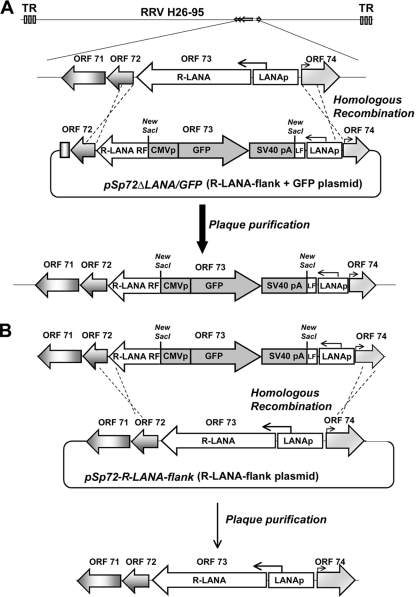
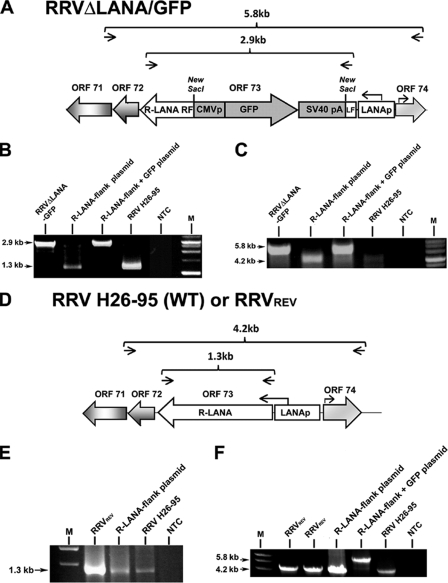
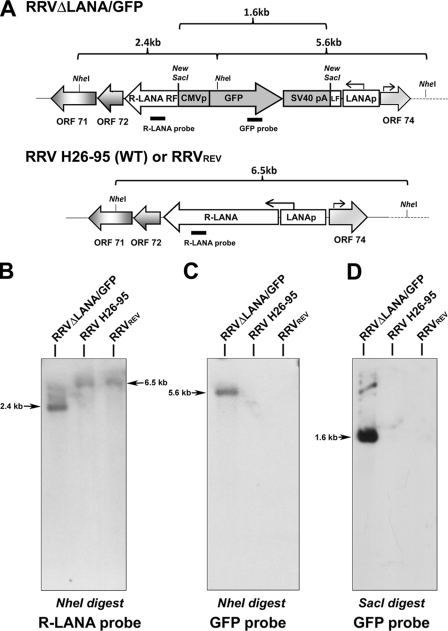
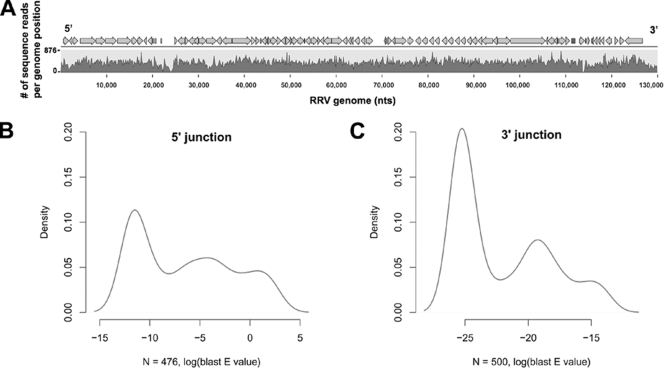
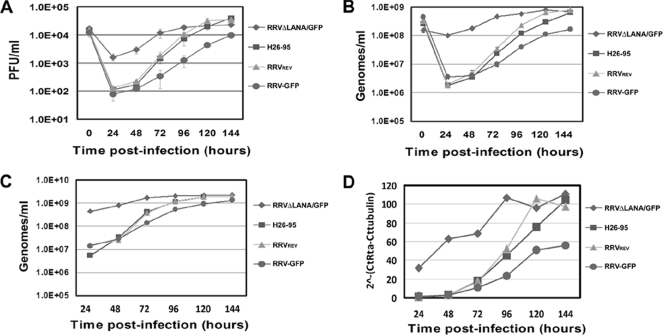
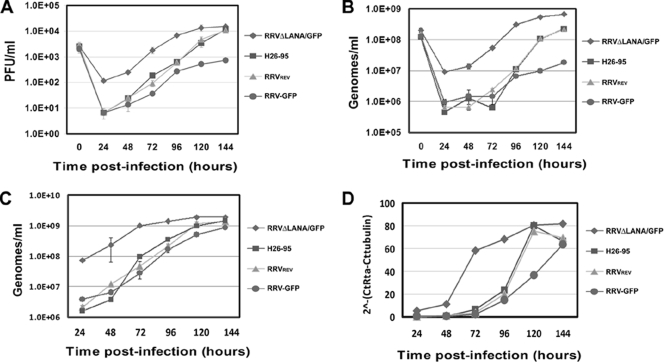
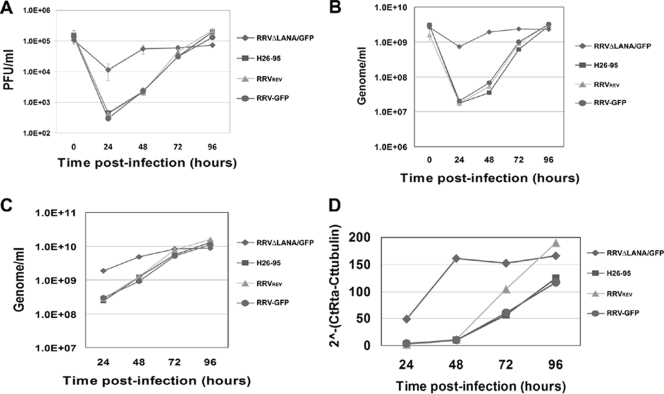
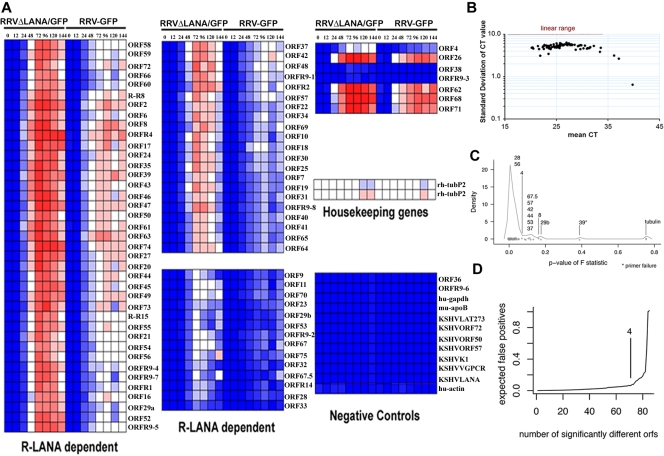
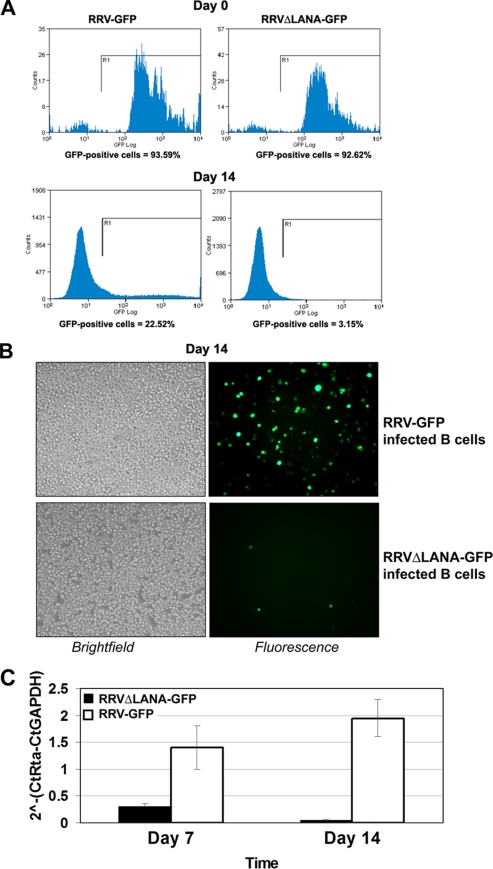
Rhesus monkey rhadinovirus (RRV) is a gammaherpesvirus that is closely related to human Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8). RRV is the closest relative to KSHV that has a fully sequenced genome and serves as an in vitro and an in vivo model system for KSHV. The latency-associated nuclear antigen (LANA) protein of both KSHV and RRV plays key roles in the establishment and maintenance of these herpesviruses. We have constructed a RRV recombinant virus (RRVDeltaLANA/GFP) in which the RRV LANA open reading frame has been disrupted with a green fluorescent protein (GFP) expression cassette generated by homologous recombination. The integrity of the recombinant virus was confirmed by diagnostic PCR, restriction digestion, Southern blot analysis, and whole-genome sequencing. We compared the single-step and multistep replication kinetics of RRVDeltaLANA/GFP, RRV-GFP, wild-type (WT) RRV H26-95, and a revertant virus using traditional plaque assays, as well as real-time quantitative PCR-based genome quantification assays. The RRVDeltaLANA/GFP recombinant virus exhibited significantly higher lytic replicative properties compared to RRV-GFP, WT RRV, or the revertant virus. This was observed upon de novo infection and in the absence of chemical inducers such as phorbol esters. In addition, by using a quantitative real-time PCR-based viral array, we are the first to report differences in global viral gene expression between WT and recombinant viruses. The RRVDeltaLANA/GFP virus displayed increased lytic gene transcription at all time points postinfection compared to RRV-GFP. Moreover, we also examined several cellular genes that are known to be repressed by KSHV LANA and report that these genes are derepressed during de novo lytic infection with the RRVDeltaLANA/GFP virus compared to RRV-GFP. Finally, we also demonstrate that the RRVDeltaLANA/GFP virus fails to establish latency in B cells, as measured by the loss of GFP-positive cells and intracellular viral genomes.
Figures
Similar articles
-
Murine Gammaherpesvirus 68 Expressing Kaposi Sarcoma-Associated Herpesvirus Latency-Associated Nuclear Antigen (LANA) Reveals both Functional Conservation and Divergence in LANA Homologs.J Virol. 2017 Sep 12;91(19):e00992-17. doi: 10.1128/JVI.00992-17. Print 2017 Oct 1. J Virol. 2017. PMID: 28747501 Free PMC article.
-
Rhesus monkey rhadinovirus (RRV): construction of a RRV-GFP recombinant virus and development of assays to assess viral replication.Virology. 2003 Jul 20;312(1):122-34. doi: 10.1016/s0042-6822(03)00195-8. Virology. 2003. PMID: 12890626
-
The latency-associated nuclear antigen of rhesus monkey rhadinovirus inhibits viral replication through repression of Orf50/Rta transcriptional activation.J Virol. 2005 Mar;79(5):3127-38. doi: 10.1128/JVI.79.5.3127-3138.2005. J Virol. 2005. PMID: 15709032 Free PMC article.
-
Rhesus monkey rhadinovirus: a model for the study of KSHV.Curr Top Microbiol Immunol. 2007;312:43-69. doi: 10.1007/978-3-540-34344-8_2. Curr Top Microbiol Immunol. 2007. PMID: 17089793 Review.
-
KSHV Genome Replication and Maintenance in Latency.Adv Exp Med Biol. 2018;1045:299-320. doi: 10.1007/978-981-10-7230-7_14. Adv Exp Med Biol. 2018. PMID: 29896673 Review.
Cited by
-
Sequence variability in clinical and laboratory isolates of herpes simplex virus 1 reveals new mutations.J Virol. 2010 May;84(10):5303-13. doi: 10.1128/JVI.00312-10. Epub 2010 Mar 10. J Virol. 2010. PMID: 20219902 Free PMC article.
-
Genome-wide histone acetylation profiling of Herpesvirus saimiri in human T cells upon induction with a histone deacetylase inhibitor.J Virol. 2011 Jun;85(11):5456-64. doi: 10.1128/JVI.00164-11. Epub 2011 Mar 23. J Virol. 2011. PMID: 21430050 Free PMC article.
-
An essential role for γ-herpesvirus latency-associated nuclear antigen homolog in an acute lymphoproliferative disease of cattle.Proc Natl Acad Sci U S A. 2013 May 21;110(21):E1933-42. doi: 10.1073/pnas.1216531110. Epub 2013 Apr 29. Proc Natl Acad Sci U S A. 2013. PMID: 23630278 Free PMC article.
-
Phosphorylation of the chromatin binding domain of KSHV LANA.PLoS Pathog. 2012;8(10):e1002972. doi: 10.1371/journal.ppat.1002972. Epub 2012 Oct 18. PLoS Pathog. 2012. PMID: 23093938 Free PMC article.
-
The latency-associated nuclear antigen, a multifunctional protein central to Kaposi's sarcoma-associated herpesvirus latency.Future Microbiol. 2011 Dec;6(12):1399-413. doi: 10.2217/fmb.11.137. Future Microbiol. 2011. PMID: 22122438 Free PMC article. Review.
References
-
- Alexander, L., L. Denekamp, A. Knapp, M. R. Auerbach, B. Damania, and R. C. Desrosiers. 2000. The primary sequence of rhesus monkey rhadinovirus isolate 26-95: sequence similarities to Kaposi's sarcoma-associated herpesvirus and rhesus monkey rhadinovirus isolate 17577. J. Virol. 74:3388-3398. - PMC - PubMed
-
- An, F. Q., N. Compitello, E. Horwitz, M. Sramkoski, E. S. Knudsen, and R. Renne. 2005. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus modulates cellular gene expression and protects lymphoid cells from p16 INK4A-induced cell cycle arrest. J. Biol. Chem. 280:3862-3874. - PubMed
-
- An, J., Y. Sun, and M. B. Rettig. 2004. Transcriptional coactivation of c-Jun by the KSHV-encoded LANA. Blood 103:222-228. - PubMed
-
- Bajaj, B. G., S. C. Verma, K. Lan, M. A. Cotter, Z. L. Woodman, and E. S. Robertson. 2006. KSHV encoded LANA upregulates Pim-1 and is a substrate for its kinase activity. Virology 351:18-28. - PubMed
-
- Ballestas, M. E., P. A. Chatis, and K. M. Kaye. 1999. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 284:641-644. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
