

CSF-1 signals directly to renal tubular epithelial cells to mediate repair in mice
- PMID: 19587445
- PMCID: PMC2719924
- DOI: 10.1172/JCI39087
CSF-1 signals directly to renal tubular epithelial cells to mediate repair in mice
Abstract
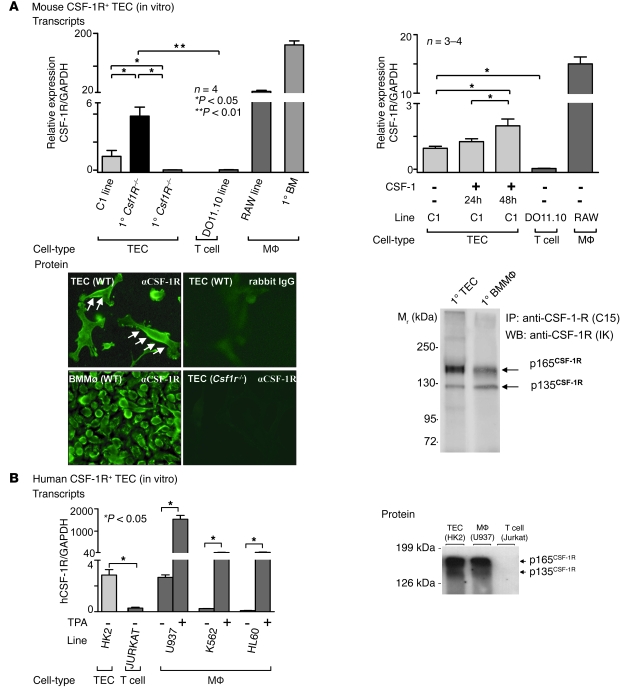
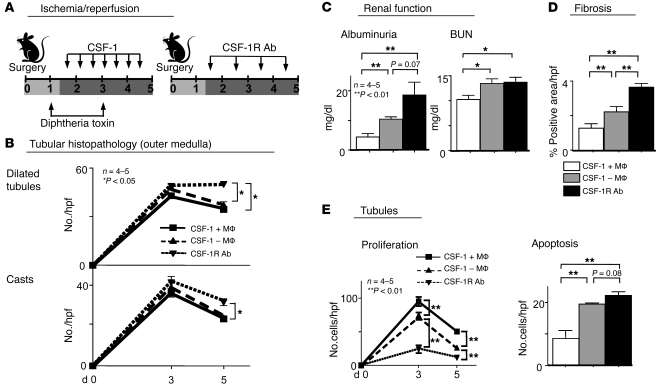
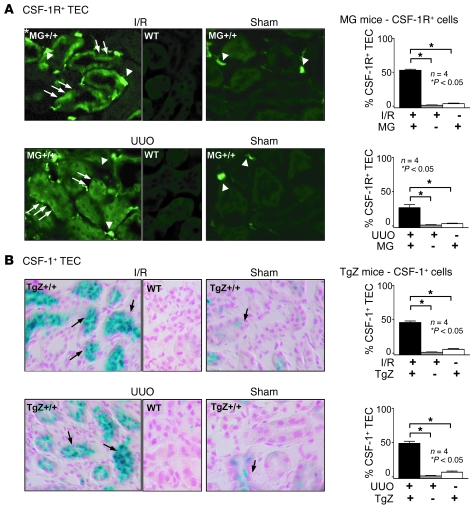
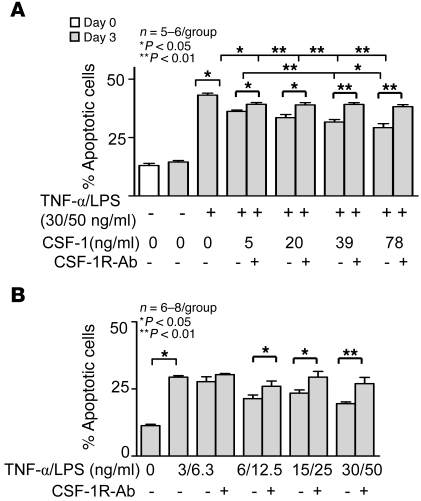
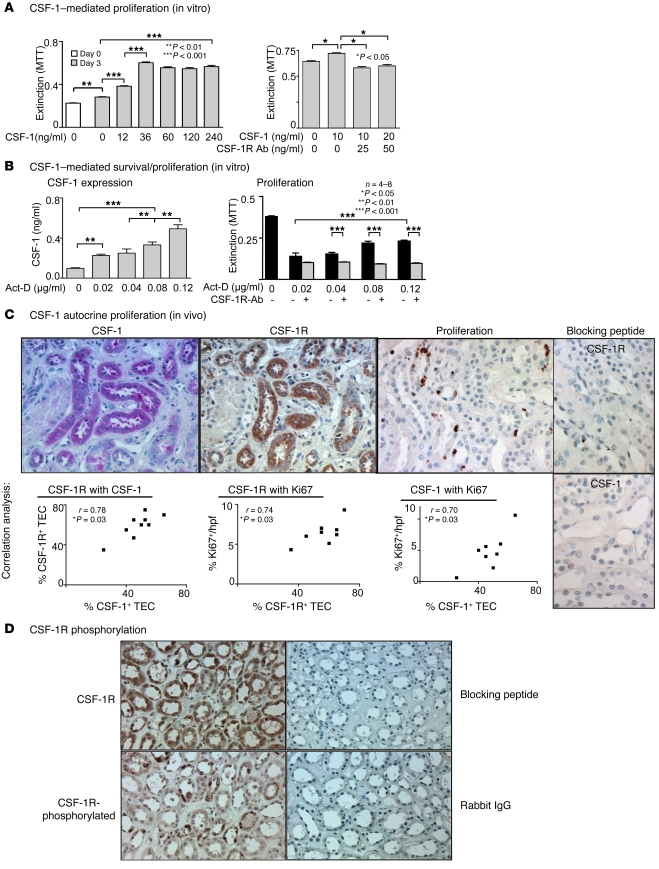
Tubular damage following ischemic renal injury is often reversible, and tubular epithelial cell (TEC) proliferation is a hallmark of tubular repair. Macrophages have been implicated in tissue repair, and CSF-1, the principal macrophage growth factor, is expressed by TECs. We therefore tested the hypothesis that CSF-1 is central to tubular repair using an acute renal injury and repair model, ischemia/reperfusion (I/R). Mice injected with CSF-1 following I/R exhibited hastened healing, as evidenced by decreased tubular pathology, reduced fibrosis, and improved renal function. Notably, CSF-1 treatment increased TEC proliferation and reduced TEC apoptosis. Moreover, administration of a CSF-1 receptor-specific (CSF-1R-specific) antibody after I/R increased tubular pathology and fibrosis, suppressed TEC proliferation, and heightened TEC apoptosis. To determine the contribution of macrophages to CSF-1-dependent renal repair, we assessed the effect of CSF-1 on I/R in mice in which CD11b+ cells were genetically ablated and determined that macrophages only partially accounted for CSF-1-dependent tubular repair. We found that TECs expressed the CSF-1R and that this receptor was upregulated and coexpressed with CSF-1 in TECs following renal injury in mice and humans. Furthermore, signaling via the CSF-1R stimulated proliferation and reduced apoptosis in human and mouse TECs. Taken together, these data suggest that CSF-1 mediates renal repair by both a macrophage-dependent mechanism and direct autocrine/paracrine action on TECs.
Figures

Similar articles
-
Trib1 Contributes to Recovery From Ischemia/Reperfusion-Induced Acute Kidney Injury by Regulating the Polarization of Renal Macrophages.Front Immunol. 2020 Mar 20;11:473. doi: 10.3389/fimmu.2020.00473. eCollection 2020. Front Immunol. 2020. PMID: 32265926 Free PMC article.
-
Reduced macrophage recruitment, proliferation, and activation in colony-stimulating factor-1-deficient mice results in decreased tubular apoptosis during renal inflammation.J Immunol. 2003 Mar 15;170(6):3254-62. doi: 10.4049/jimmunol.170.6.3254. J Immunol. 2003. PMID: 12626584
-
Chronicity following ischaemia-reperfusion injury depends on tubular-macrophage crosstalk involving two tubular cell-derived CSF-1R activators: CSF-1 and IL-34.Nephrol Dial Transplant. 2016 Sep;31(9):1409-16. doi: 10.1093/ndt/gfw026. Epub 2016 Mar 24. Nephrol Dial Transplant. 2016. PMID: 27190368 Review.
-
Macrophage growth factors introduced into the kidney initiate renal injury.Mol Med. 1996 May;2(3):297-312. Mol Med. 1996. PMID: 8784783 Free PMC article.
-
Metabolic Flexibility and Innate Immunity in Renal Ischemia Reperfusion Injury: The Fine Balance Between Adaptive Repair and Tissue Degeneration.Front Immunol. 2020 Jul 7;11:1346. doi: 10.3389/fimmu.2020.01346. eCollection 2020. Front Immunol. 2020. PMID: 32733450 Free PMC article. Review.
Cited by
-
Immunity and inflammation in diabetic kidney disease: translating mechanisms to biomarkers and treatment targets.Am J Physiol Renal Physiol. 2017 Apr 1;312(4):F716-F731. doi: 10.1152/ajprenal.00314.2016. Epub 2016 Aug 24. Am J Physiol Renal Physiol. 2017. PMID: 27558558 Free PMC article. Review.
-
Biomarkers for childhood-onset systemic lupus erythematosus.Curr Rheumatol Rep. 2015 Jan;17(1):471. doi: 10.1007/s11926-014-0471-2. Curr Rheumatol Rep. 2015. PMID: 25475594 Free PMC article. Review.
-
Reno-protective effect of IL-34 inhibition on cisplatin-induced nephrotoxicity in mice.PLoS One. 2021 Jan 11;16(1):e0245340. doi: 10.1371/journal.pone.0245340. eCollection 2021. PLoS One. 2021. PMID: 33428678 Free PMC article.
-
The effect of ionizing radiation on testicular interstitial stromal cells.Reprod Med Biol. 2025 Mar 19;24(1):e12639. doi: 10.1002/rmb2.12639. eCollection 2025 Jan-Dec. Reprod Med Biol. 2025. PMID: 40110174 Free PMC article.
-
Cloning and expression of porcine Colony Stimulating Factor-1 (CSF-1) and Colony Stimulating Factor-1 Receptor (CSF-1R) and analysis of the species specificity of stimulation by CSF-1 and Interleukin 34.Cytokine. 2012 Dec;60(3):793-805. doi: 10.1016/j.cyto.2012.08.008. Epub 2012 Sep 10. Cytokine. 2012. PMID: 22974529 Free PMC article.
References
-
- Witzgall R., Brown D., Schwarz C., Bonventre J.V. Localization of proliferating cell nuclear antigen, vimentin, c-Fos, and clusterin in the postischemic kidney. Evidence for a heterogenous genetic response among nephron segments, and a large pool of mitotically active and dedifferentiated cells. J. Clin. Invest. 1994;93:2175–2188. doi: 10.1172/JCI117214. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
