

Genomic profiling distinguishes familial multiple and sporadic multiple meningiomas
- PMID: 19589153
- PMCID: PMC2716362
- DOI: 10.1186/1755-8794-2-42
Genomic profiling distinguishes familial multiple and sporadic multiple meningiomas
Abstract
Background: Meningiomas may occur either as familial tumors in two distinct disorders, familial multiple meningioma and neurofibromatosis 2 (NF2), or sporadically, as either single or multiple tumors in individuals with no family history. Meningiomas in NF2 and approximately 60% of sporadic meningiomas involve inactivation of the NF2 locus, encoding the tumor suppressor merlin on chromosome 22q. This study was undertaken to establish whether genomic profiling could distinguish familial multiple meningiomas from sporadic solitary and sporadic multiple meningiomas.
Methods: We compared 73 meningiomas presenting as sporadic solitary (64), sporadic multiple (5) and familial multiple (4) tumors using genomic profiling by array comparative genomic hybridization (array CGH).
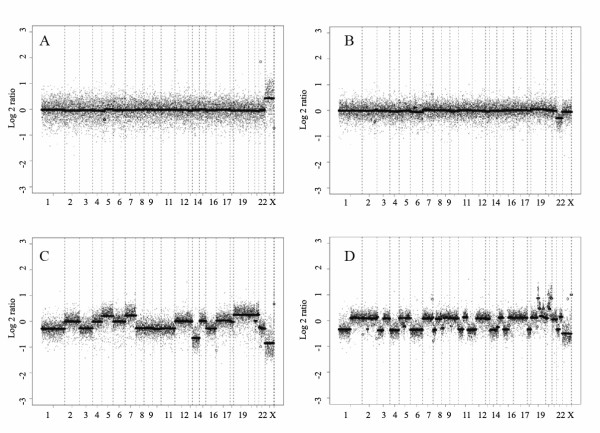
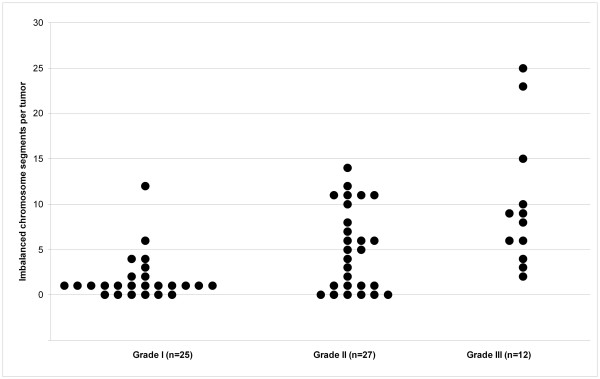
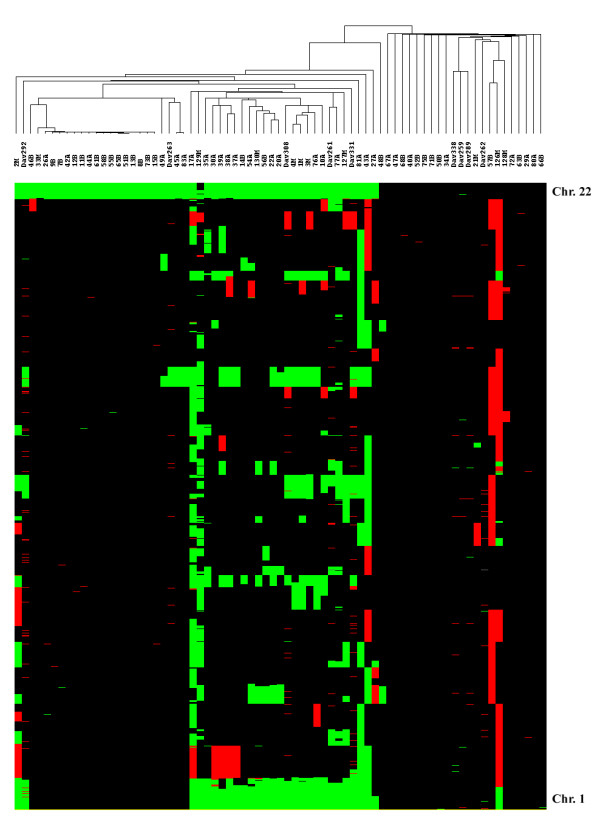
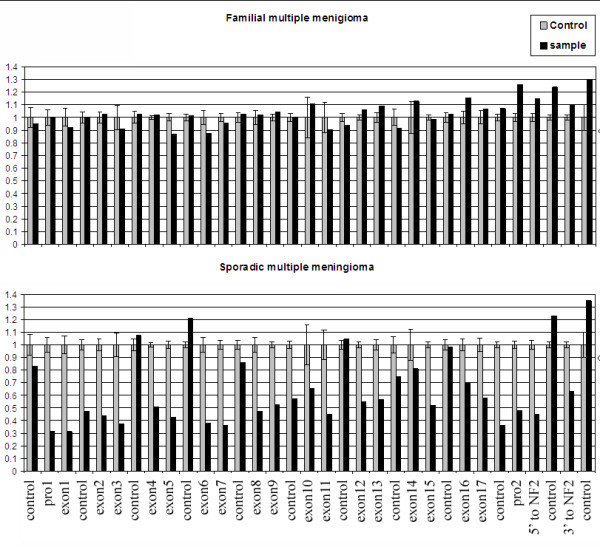
Results: Sporadic solitary meningiomas revealed genomic rearrangements consistent with at least two mechanisms of tumor initiation, as unsupervised cluster analysis readily distinguished tumors with chromosome 22 deletion (associated with loss of the NF2 tumor suppressor) from those without chromosome 22 deletion. Whereas sporadic meningiomas without chromosome 22 loss exhibited fewer chromosomal imbalance events overall, tumors with chromosome 22 deletion further clustered into two major groups that largely, though not perfectly, matched with their benign (WHO Grade I) or advanced (WHO Grades II and III) histological grade, with the latter exhibiting a significantly greater degree of genomic imbalance (P < 0.001). Sporadic multiple meningiomas showed a frequency of genomic imbalance events comparable to the atypical grade solitary tumors. By contrast, familial multiple meningiomas displayed no imbalances, supporting a distinct mechanism for the origin for these tumors.
Conclusion: Genomic profiling can provide an unbiased adjunct to traditional meningioma classification and provides a basis for exploring the different genetic underpinnings of tumor initiation and progression. Most importantly, the striking difference observed between sporadic and familial multiple meningiomas indicates that genomic profiling can provide valuable information for differential diagnosis of subjects with multiple meningiomas and for considering the risk for tumor occurrence in their family members.
Figures
References
-
- CBTRUS . Statistical Report: Primary Brain Tumors in the United States, 2000–2004. Published by the Central Brain Tumor Registry of the United States.; USA; 2008.
-
- Evans DG, Huson SM, Donnai D, Neary W, Blair V, Newton V, Harris R. A clinical study of type 2 neurofibromatosis. Q J Med. 1992;84:603–618. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
