

The role of liver-derived insulin-like growth factor-I
- PMID: 19589948
- PMCID: PMC2759708
- DOI: 10.1210/er.2009-0010
The role of liver-derived insulin-like growth factor-I
Abstract
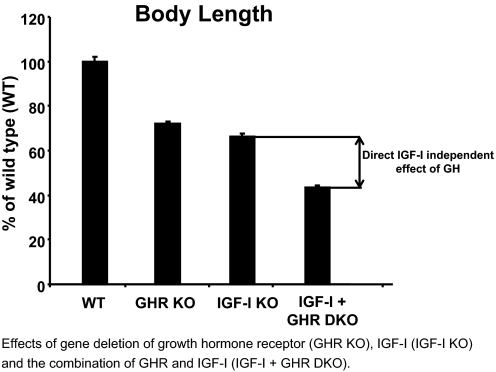
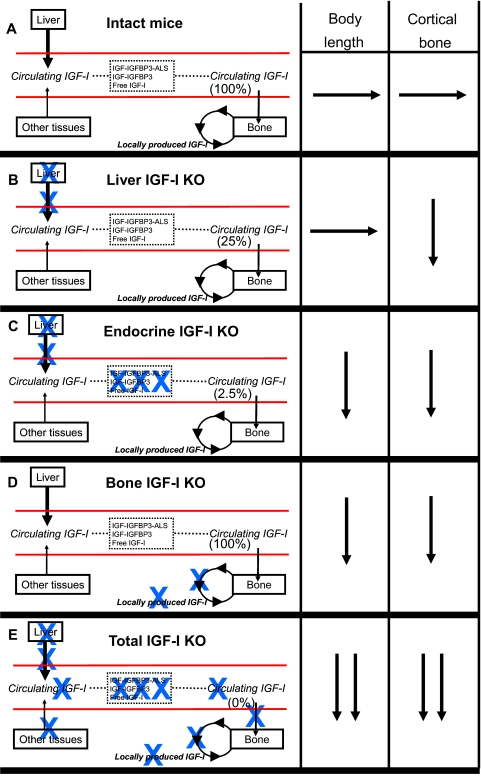
IGF-I is expressed in virtually every tissue of the body, but with much higher expression in the liver than in any other tissue. Studies using mice with liver-specific IGF-I knockout have demonstrated that liver-derived IGF-I, constituting a major part of circulating IGF-I, is an important endocrine factor involved in a variety of physiological and pathological processes. Detailed studies comparing the impact of liver-derived IGF-I and local bone-derived IGF-I demonstrate that both sources of IGF-I can stimulate longitudinal bone growth. We propose here that liver-derived circulating IGF-I and local bone-derived IGF-I to some extent have overlapping growth-promoting effects and might have the capacity to replace each other (= redundancy) in the maintenance of normal longitudinal bone growth. Importantly, and in contrast to the regulation of longitudinal bone growth, locally derived IGF-I cannot replace (= lack of redundancy) liver-derived IGF-I for the regulation of a large number of other parameters including GH secretion, cortical bone mass, kidney size, prostate size, peripheral vascular resistance, spatial memory, sodium retention, insulin sensitivity, liver size, sexually dimorphic liver functions, and progression of some tumors. It is clear that a major role of liver-derived IGF-I is to regulate GH secretion and that some, but not all, of the phenotypes in the liver-specific IGF-I knockout mice are indirect, mediated via the elevated GH levels. All of the described multiple endocrine effects of liver-derived IGF-I should be considered in the development of possible novel treatment strategies aimed at increasing or reducing endocrine IGF-I activity.
Figures







References
-
- Daughaday WH, Rotwein P 1989 Insulin-like growth factors I and II. Peptide, messenger ribonucleic acid and gene structures, serum, and tissue concentrations. Endocr Rev 10:68–91 - PubMed
-
- Isaksson OG, Lindahl A, Nilsson A, Isgaard J 1987 Mechanism of the stimulatory effect of growth hormone on longitudinal bone growth. Endocr Rev 8:426–438 - PubMed
-
- Ohlsson C, Bengtsson BA, Isaksson OG, Andreassen TT, Slootweg MC 1998 Growth hormone and bone. Endocr Rev 19:55–79 - PubMed
-
- Le Roith D, Bondy C, Yakar S, Liu JL, Butler A 2001 The somatomedin hypothesis: 2001. Endocr Rev 22:53–74 - PubMed
-
- Samani AA, Yakar S, LeRoith D, Brodt P 2007 The role of the IGF system in cancer growth and metastasis: overview and recent insights. Endocr Rev 28:20–47 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
