

Pathogenesis, diagnosis, and management of primary antibody deficiencies and infections
- PMID: 19597006
- PMCID: PMC2708392
- DOI: 10.1128/CMR.00001-09
Pathogenesis, diagnosis, and management of primary antibody deficiencies and infections
Abstract
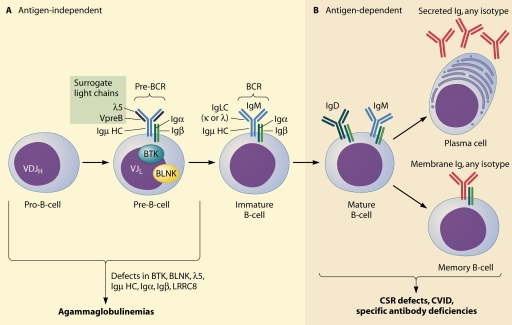
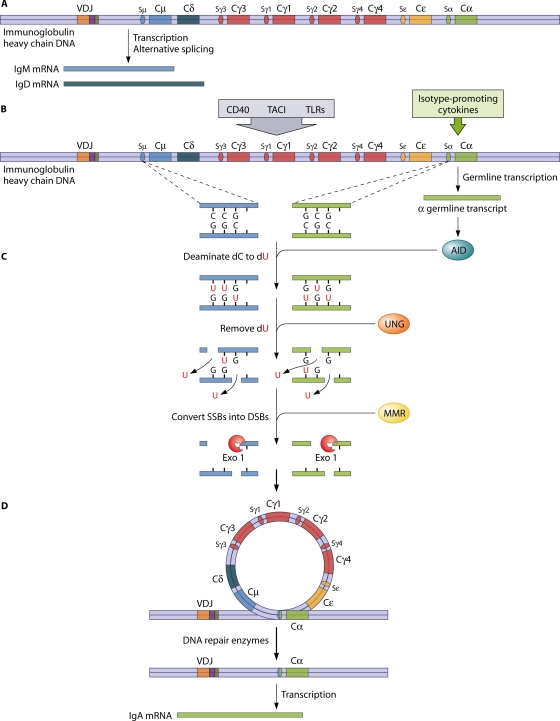
Primary antibody deficiencies are the most common primary immunodeficiency diseases. They are a heterogeneous group of disorders with various degrees of dysfunctional antibody production resulting from a disruption of B-cell differentiation at different stages. While there has been tremendous recent progress in the understanding of some of these disorders, the etiology remains unknown for the majority of patients. As there is a large spectrum of underlying defects, the age at presentation varies widely, and the clinical manifestations range from an almost complete absence of B cells and serum immunoglobulins to selectively impaired antibody responses to specific antigens with normal total serum immunoglobulin concentrations. However, all of these disorders share an increased susceptibility to infections, affecting predominantly the respiratory tract. A delay of appropriate treatment for some diseases can result in serious complications related to infections, while timely diagnosis and adequate therapy can significantly decrease morbidity and increase life expectancy and quality of life.
Figures
References
-
- Abele-Horn, M., C. Wolff, P. Dressel, A. Zimmermann, W. Vahlensieck, F. Pfaff, and G. Ruckdeschel. 1996. Polymerase chain reaction versus culture for detection of Ureaplasma urealyticum and Mycoplasma hominis in the urogenital tract of adults and the respiratory tract of newborns. Eur. J. Clin. Microbiol. Infect. Dis. 15:595-598. - PubMed
-
- Alaswad, B., and P. Brosnan. 2000. The association of celiac disease, diabetes mellitus type 1, hypothyroidism, chronic liver disease, and selective IgA deficiency. Clin. Pediatr. (Philadelphia) 39:229-231. - PubMed
-
- al Ghonaium, A., J. B. Ziegler, and D. Tridgell. 1996. Bilateral chronic conjunctivitis and corneal scarring in a boy with X-linked hypogammaglobulinaemia. J. Paediatr. Child. Health 32:463-465. - PubMed
-
- Alper, C. A. 1995. The human immune response to hepatitis B surface antigen. Exp. Clin. Immunogenet. 12:171-181. - PubMed
-
- Alt, F. W., T. K. Blackwell, and G. D. Yancopoulos. 1987. Development of the primary antibody repertoire. Science 238:1079-1087. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
