

Identification of deleterious mutations within three human genomes
- PMID: 19602639
- PMCID: PMC2752137
- DOI: 10.1101/gr.092619.109
Identification of deleterious mutations within three human genomes
Abstract
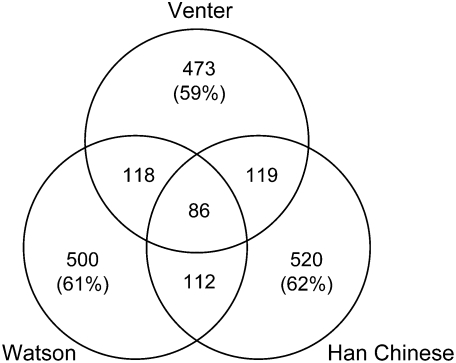
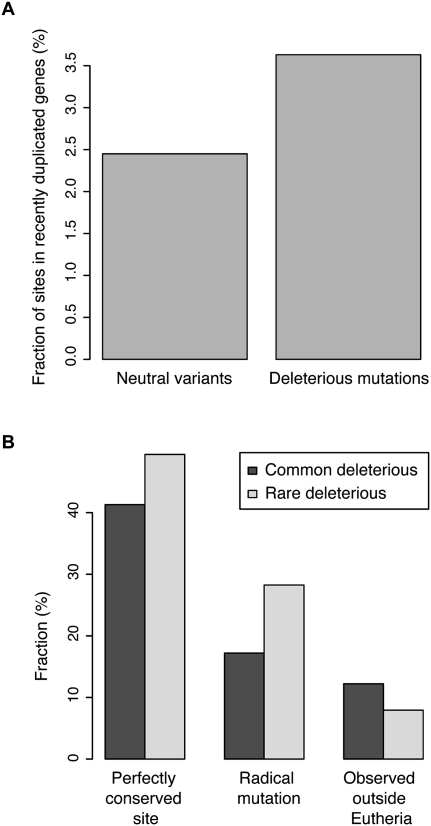
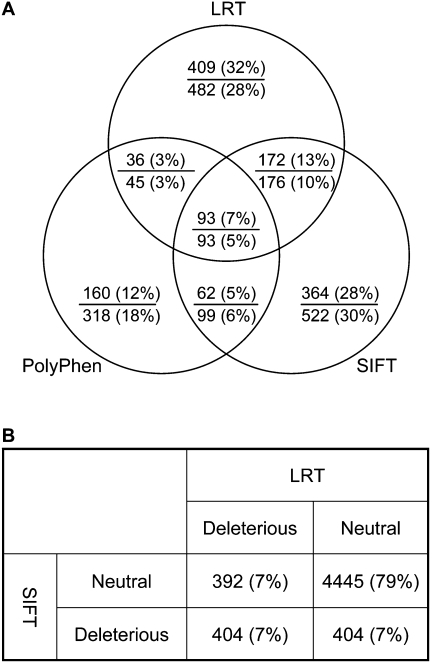
Each human carries a large number of deleterious mutations. Together, these mutations make a significant contribution to human disease. Identification of deleterious mutations within individual genome sequences could substantially impact an individual's health through personalized prevention and treatment of disease. Yet, distinguishing deleterious mutations from the massive number of nonfunctional variants that occur within a single genome is a considerable challenge. Using a comparative genomics data set of 32 vertebrate species we show that a likelihood ratio test (LRT) can accurately identify a subset of deleterious mutations that disrupt highly conserved amino acids within protein-coding sequences, which are likely to be unconditionally deleterious. The LRT is also able to identify known human disease alleles and performs as well as two commonly used heuristic methods, SIFT and PolyPhen. Application of the LRT to three human genomes reveals 796-837 deleterious mutations per individual, approximately 40% of which are estimated to be at <5% allele frequency. However, the overlap between predictions made by the LRT, SIFT, and PolyPhen, is low; 76% of predictions are unique to one of the three methods, and only 5% of predictions are shared across all three methods. Our results indicate that only a small subset of deleterious mutations can be reliably identified, but that this subset provides the raw material for personalized medicine.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources