

A network model of a cooperative genetic landscape in brain tumors
- PMID: 19602686
- PMCID: PMC4447713
- DOI: 10.1001/jama.2009.997
A network model of a cooperative genetic landscape in brain tumors
Abstract
Context: Gliomas, particularly glioblastomas, are among the deadliest of human tumors. Gliomas emerge through the accumulation of recurrent chromosomal alterations, some of which target yet-to-be-discovered cancer genes. A persistent question concerns the biological basis for the coselection of these alterations during gliomagenesis.
Objectives: To describe a network model of a cooperative genetic landscape in gliomas and to evaluate its clinical relevance.
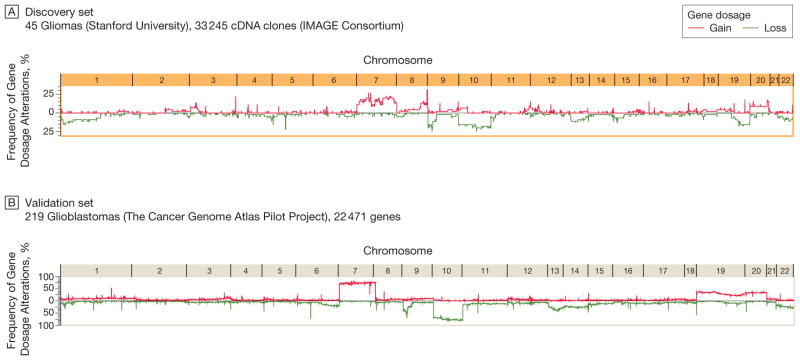
Design, setting, and patients: Multidimensional genomic profiles and clinical profiles of 501 patients with gliomas (45 tumors in an initial discovery set collected between 2001 and 2004 and 456 tumors in validation sets made public between 2006 and 2008) from multiple academic centers in the United States and The Cancer Genome Atlas Pilot Project (TCGA).
Main outcome measures: Identification of genes with coincident genetic alterations, correlated gene dosage and gene expression, and multiple functional interactions; association between those genes and patient survival.
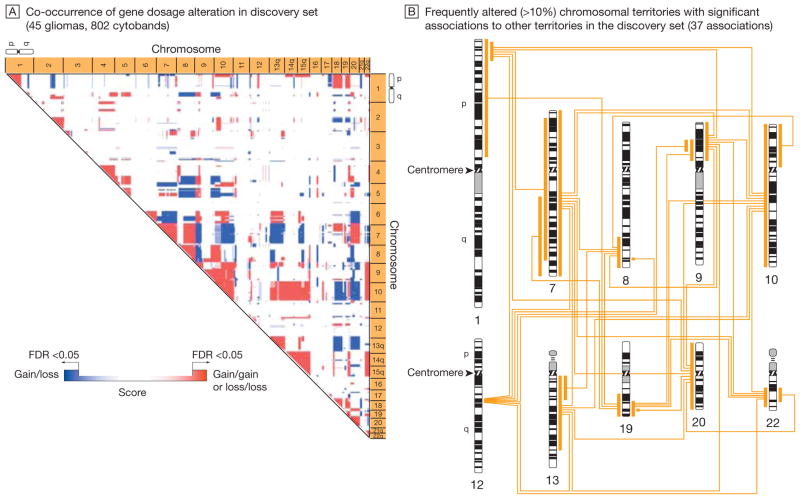
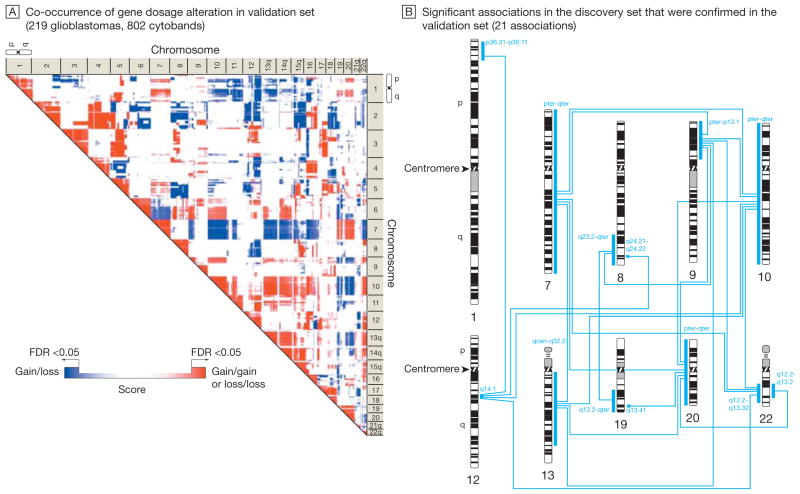
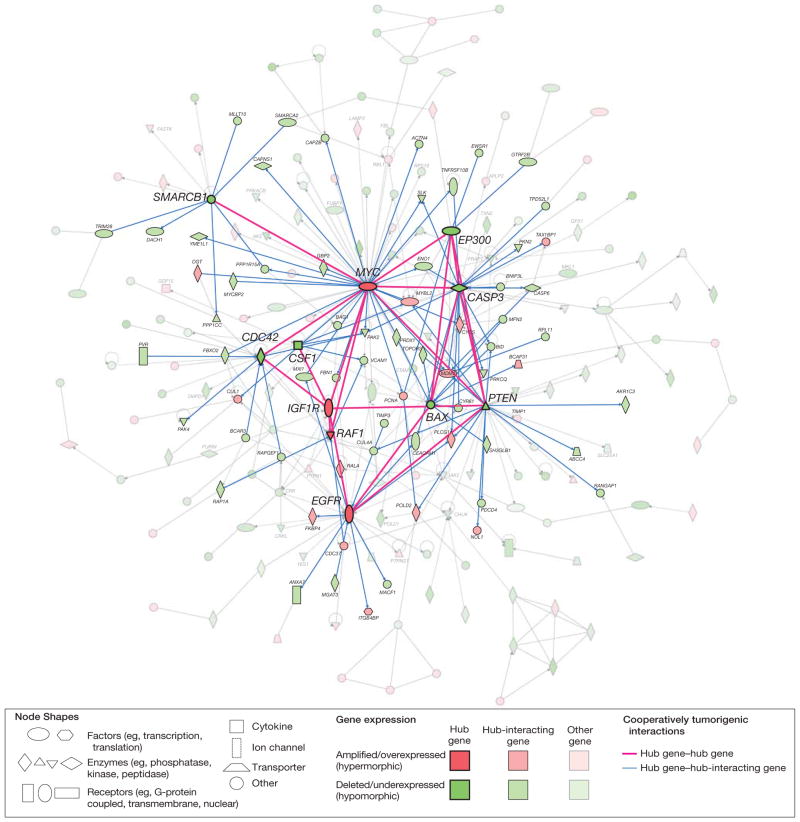
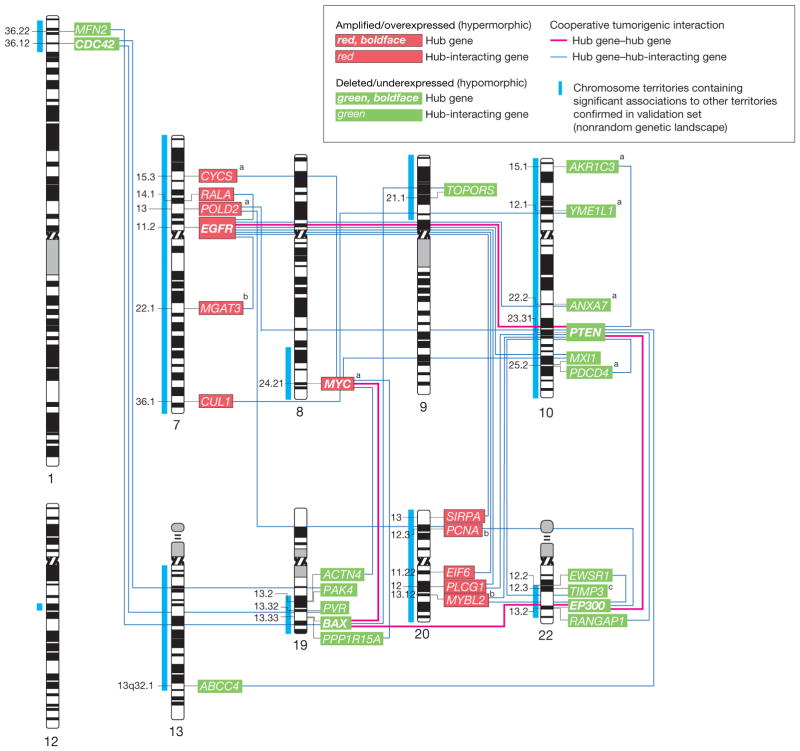
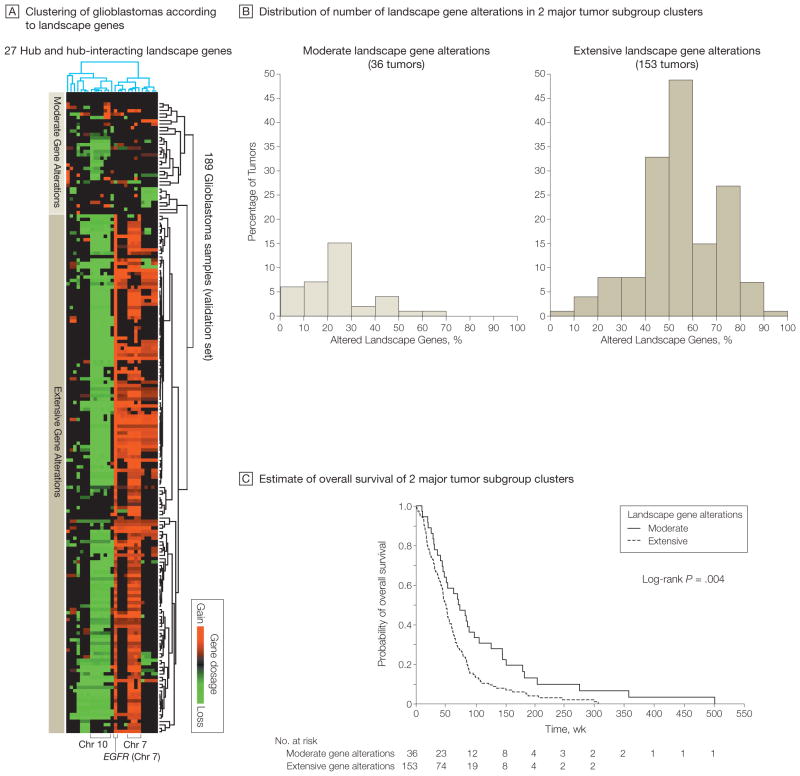
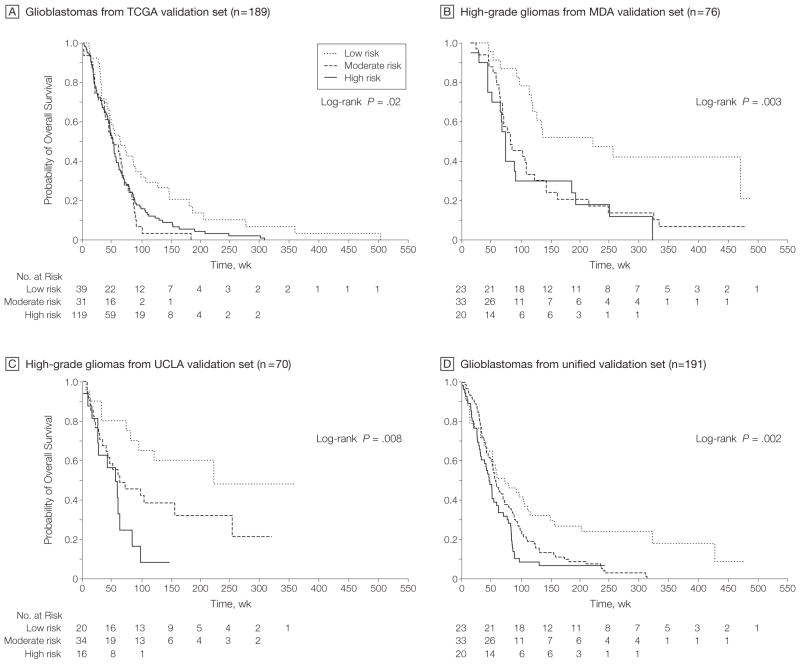
Results: Gliomas select for a nonrandom genetic landscape-a consistent pattern of chromosomal alterations-that involves altered regions ("territories") on chromosomes 1p, 7, 8q, 9p, 10, 12q, 13q, 19q, 20, and 22q (false-discovery rate-corrected P<.05). A network model shows that these territories harbor genes with putative synergistic, tumor-promoting relationships. The coalteration of the most interactive of these genes in glioblastoma is associated with unfavorable patient survival. A multigene risk scoring model based on 7 landscape genes (POLD2, CYCS, MYC, AKR1C3, YME1L1, ANXA7, and PDCD4) is associated with the duration of overall survival in 189 glioblastoma samples from TCGA (global log-rank P = .02 comparing 3 survival curves for patients with 0-2, 3-4, and 5-7 dosage-altered genes). Groups of patients with 0 to 2 (low-risk group) and 5 to 7 (high-risk group) dosage-altered genes experienced 49.24 and 79.56 deaths per 100 person-years (hazard ratio [HR], 1.63; 95% confidence interval [CI], 1.10-2.40; Cox regression model P = .02), respectively. These associations with survival are validated using gene expression data in 3 independent glioma studies, comprising 76 (global log-rank P = .003; 47.89 vs 15.13 deaths per 100 person-years for high risk vs low risk; Cox model HR, 3.04; 95% CI, 1.49-6.20; P = .002) and 70 (global log-rank P = .008; 83.43 vs 16.14 deaths per 100 person-years for high risk vs low risk; HR, 3.86; 95% CI, 1.59-9.35; P = .003) high-grade gliomas and 191 glioblastomas (global log-rank P = .002; 83.23 vs 34.16 deaths per 100 person-years for high risk vs low risk; HR, 2.27; 95% CI, 1.44-3.58; P<.001).
Conclusions: The alteration of multiple networking genes by recurrent chromosomal aberrations in gliomas deregulates critical signaling pathways through multiple, cooperative mechanisms. These mutations, which are likely due to nonrandom selection of a distinct genetic landscape during gliomagenesis, are associated with patient prognosis.
Figures
Comment in
-
One step forward toward identification of the genetic signature of glioblastomas.JAMA. 2009 Jul 15;302(3):325-6. doi: 10.1001/jama.2009.1023. JAMA. 2009. PMID: 19602695 No abstract available.
-
Malignant astrocytomas: a system disease.Arch Neurol. 2010 Mar;67(3):353-5. doi: 10.1001/archneurol.2010.19. Arch Neurol. 2010. PMID: 20212234 No abstract available.
References
-
- Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359(5):492–507. - PubMed
-
- Bredel M, Bredel C, Juric D, et al. High-resolution genome-wide mapping of genetic alterations in human glial brain tumors. Cancer Res. 2005;65(10):4088–4096. - PubMed
-
- Nigro JM, Misra A, Zhang L, et al. Integrated array-comparative genomic hybridization and expression array profiles identify clinically relevant molecular subtypes of glioblastoma. Cancer Res. 2005;65(5):1678–1686. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
