

Improved prediction of protein side-chain conformations with SCWRL4
- PMID: 19603484
- PMCID: PMC2885146
- DOI: 10.1002/prot.22488
Improved prediction of protein side-chain conformations with SCWRL4
Abstract
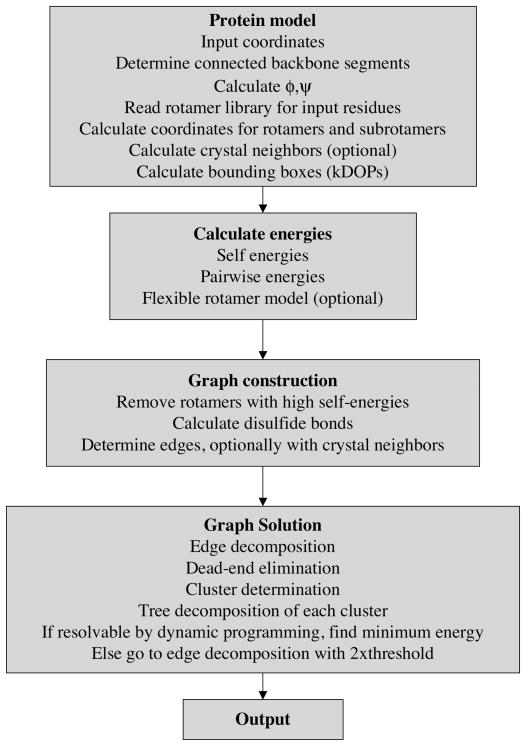
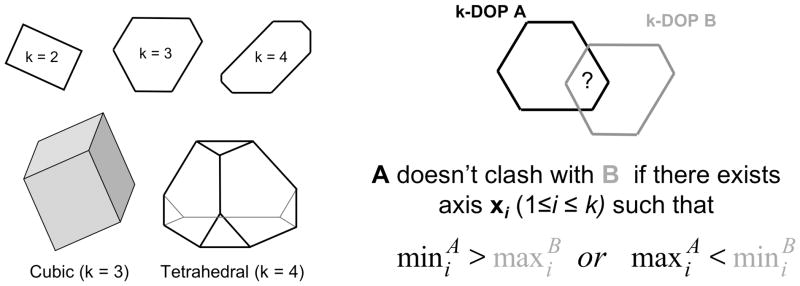
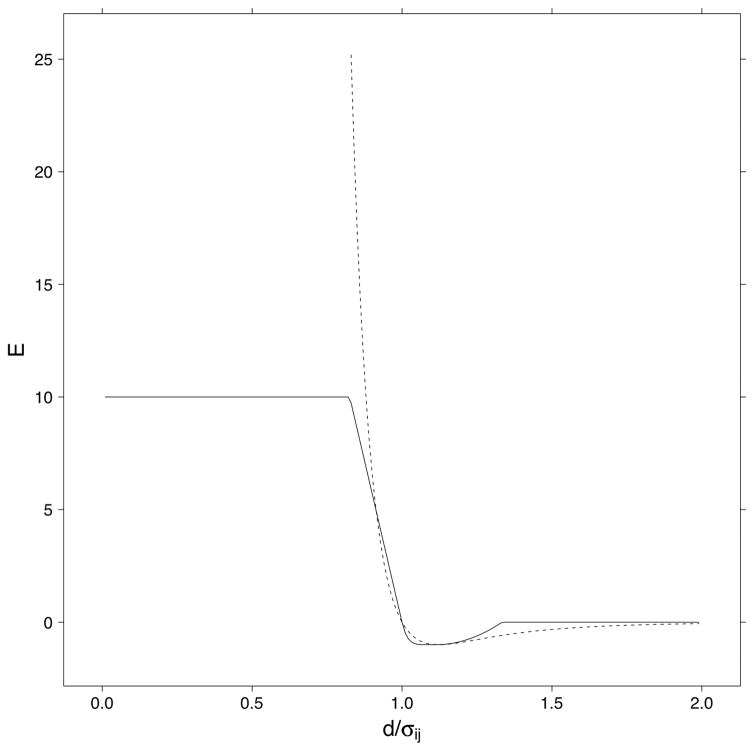
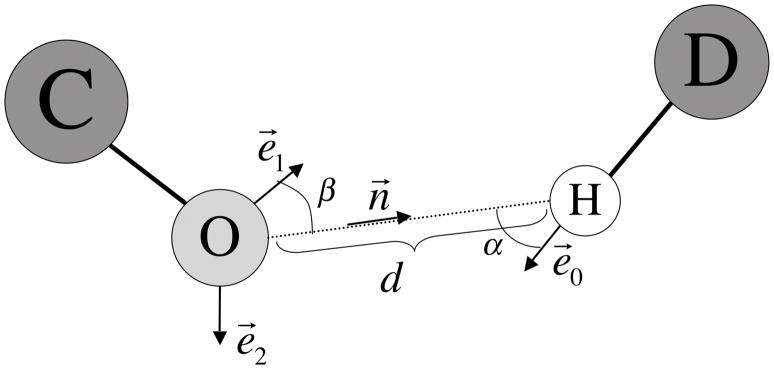
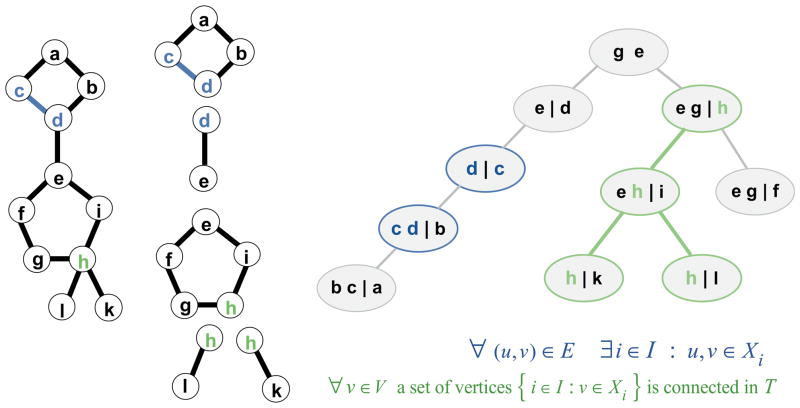
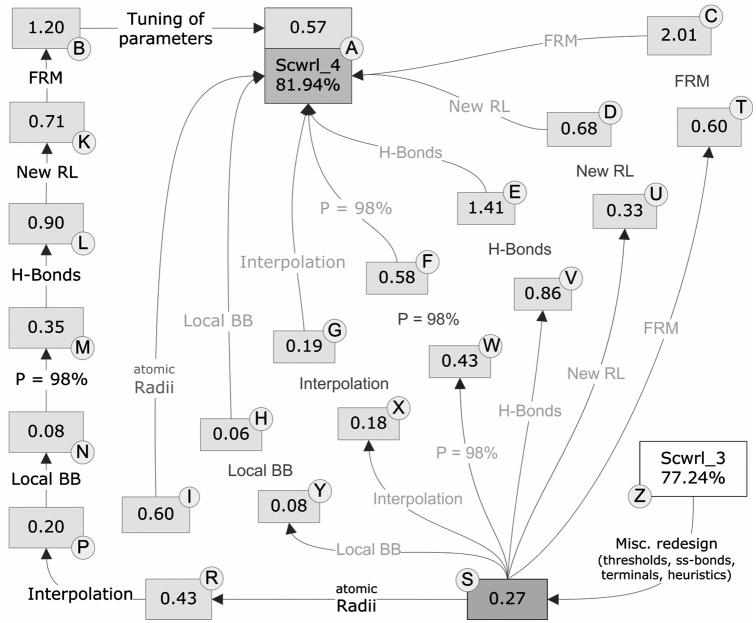
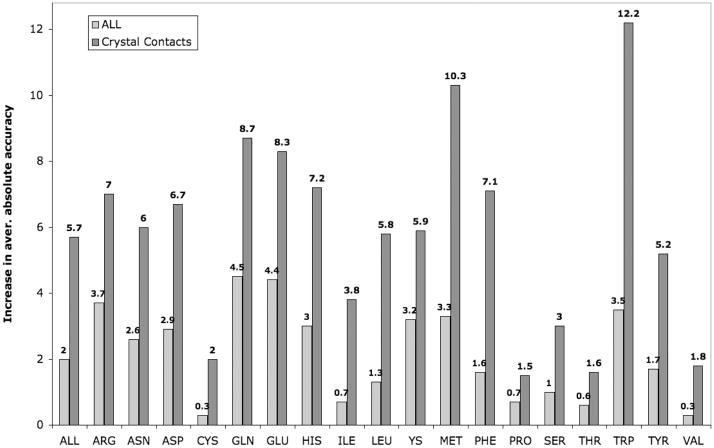
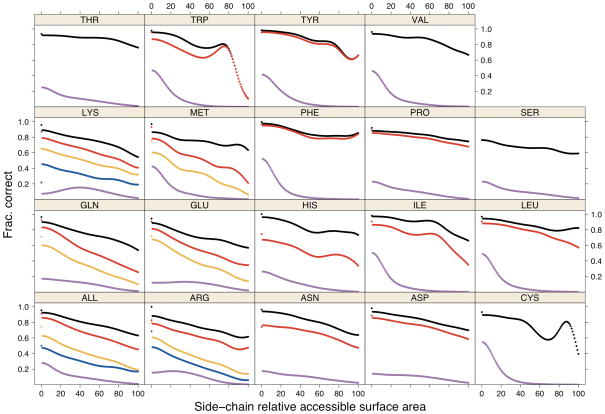
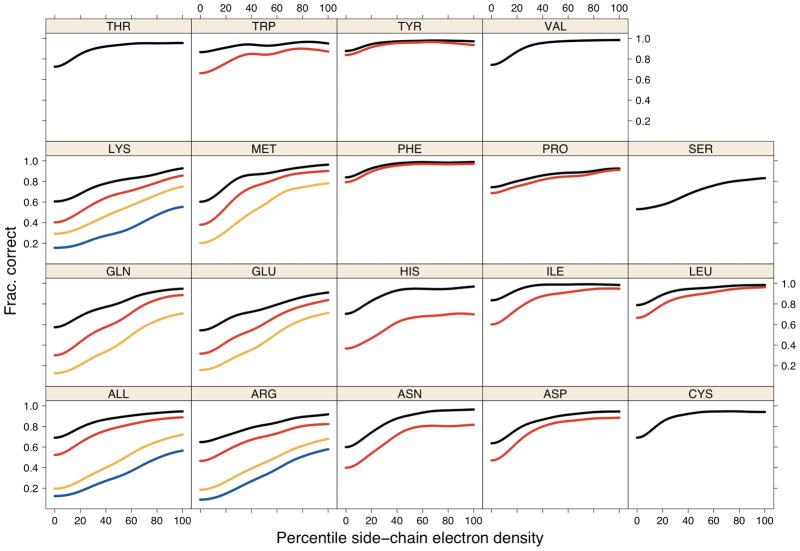
Determination of side-chain conformations is an important step in protein structure prediction and protein design. Many such methods have been presented, although only a small number are in widespread use. SCWRL is one such method, and the SCWRL3 program (2003) has remained popular because of its speed, accuracy, and ease-of-use for the purpose of homology modeling. However, higher accuracy at comparable speed is desirable. This has been achieved in a new program SCWRL4 through: (1) a new backbone-dependent rotamer library based on kernel density estimates; (2) averaging over samples of conformations about the positions in the rotamer library; (3) a fast anisotropic hydrogen bonding function; (4) a short-range, soft van der Waals atom-atom interaction potential; (5) fast collision detection using k-discrete oriented polytopes; (6) a tree decomposition algorithm to solve the combinatorial problem; and (7) optimization of all parameters by determining the interaction graph within the crystal environment using symmetry operators of the crystallographic space group. Accuracies as a function of electron density of the side chains demonstrate that side chains with higher electron density are easier to predict than those with low-electron density and presumed conformational disorder. For a testing set of 379 proteins, 86% of chi(1) angles and 75% of chi(1+2) angles are predicted correctly within 40 degrees of the X-ray positions. Among side chains with higher electron density (25-100th percentile), these numbers rise to 89 and 80%. The new program maintains its simple command-line interface, designed for homology modeling, and is now available as a dynamic-linked library for incorporation into other software programs.
2009 Wiley-Liss, Inc.
Figures
References
-
- Veenstra DL, Kollman PA. Modeling protein stability: a theoretical analysis of the stability of T4 lysozyme mutants. Protein Eng. 1997;10(7):789–807. - PubMed
-
- Gray JJ, Moughon S, Wang C, Schueler-Furman O, Kuhlman B, Rohl CA, Baker D. Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J Mol Biol. 2003;331(1):281–299. - PubMed
-
- Meiler J, Baker D. ROSETTALIGAND: protein-small molecule docking with full side-chain flexibility. Proteins. 2006;65(3):538–548. - PubMed
-
- Leach AR. Ligand docking to proteins with discrete side-chain flexibility. J Mol Biol. 1994;235(1):345–356. - PubMed
-
- Rohl CA, Strauss CE, Chivian D, Baker D. Modeling structurally variable regions in homologous proteins with rosetta. Proteins. 2004;55(3):656–677. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
