

Pathways of abnormal stress-induced Ca2+ influx into dystrophic mdx cardiomyocytes
- PMID: 19604578
- PMCID: PMC2745084
- DOI: 10.1016/j.ceca.2009.06.002
Pathways of abnormal stress-induced Ca2+ influx into dystrophic mdx cardiomyocytes
Abstract
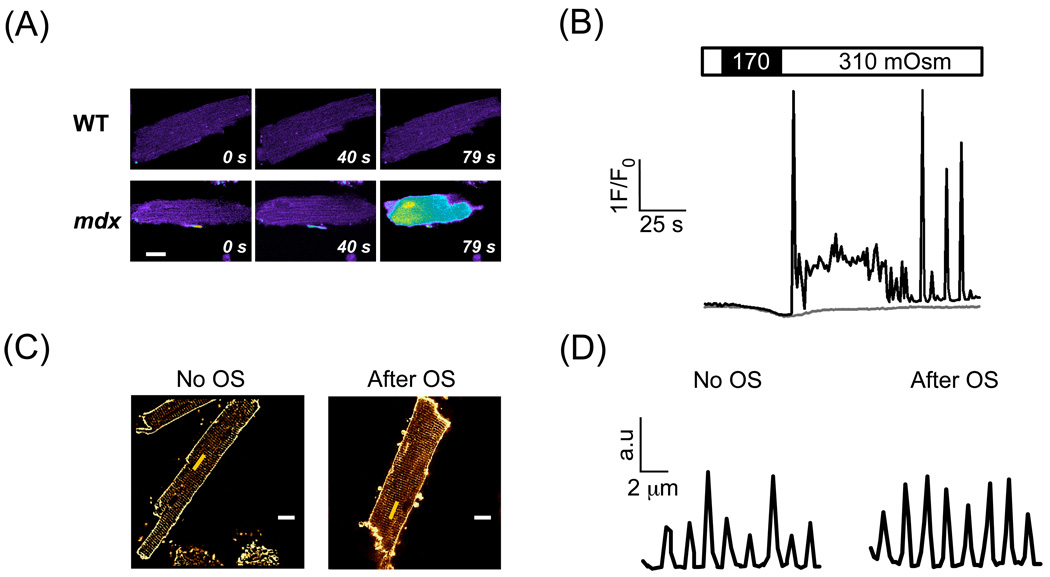
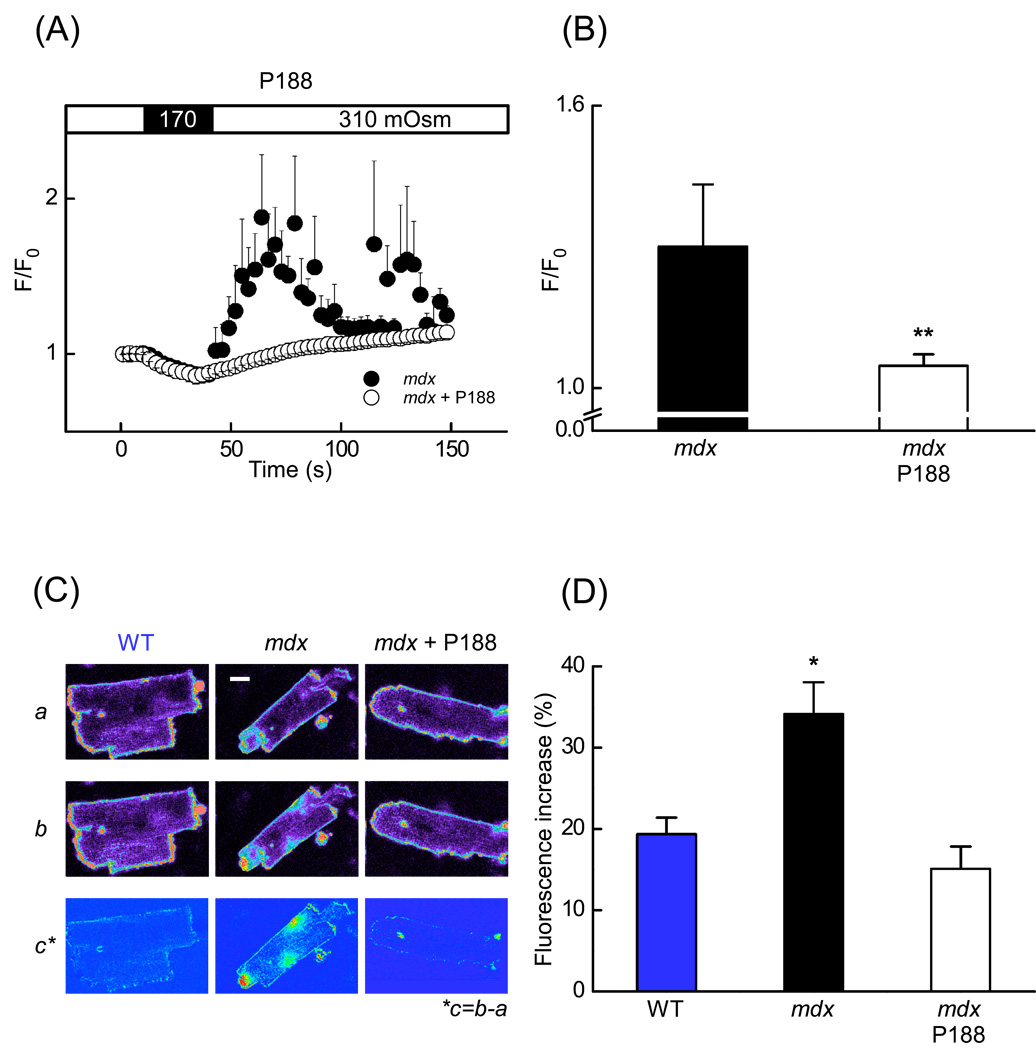
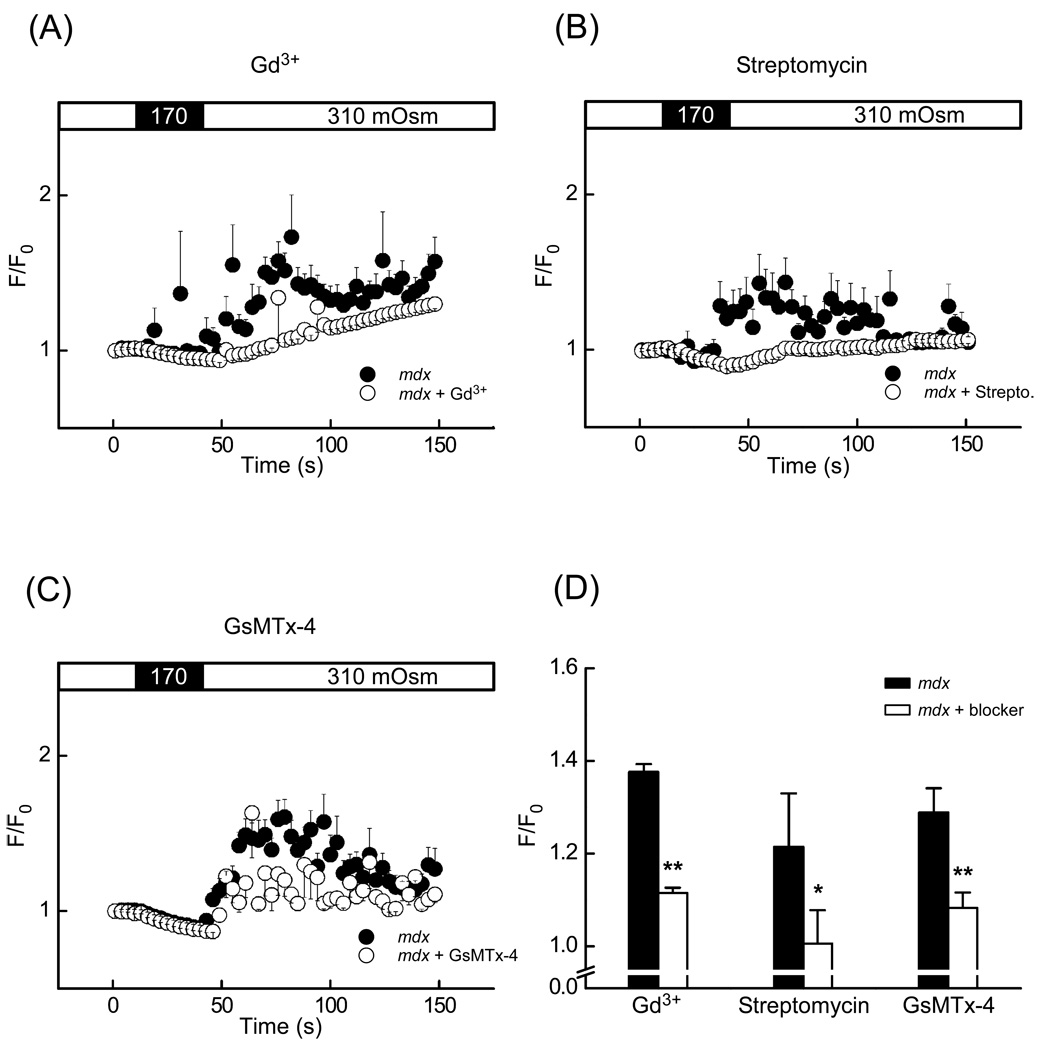
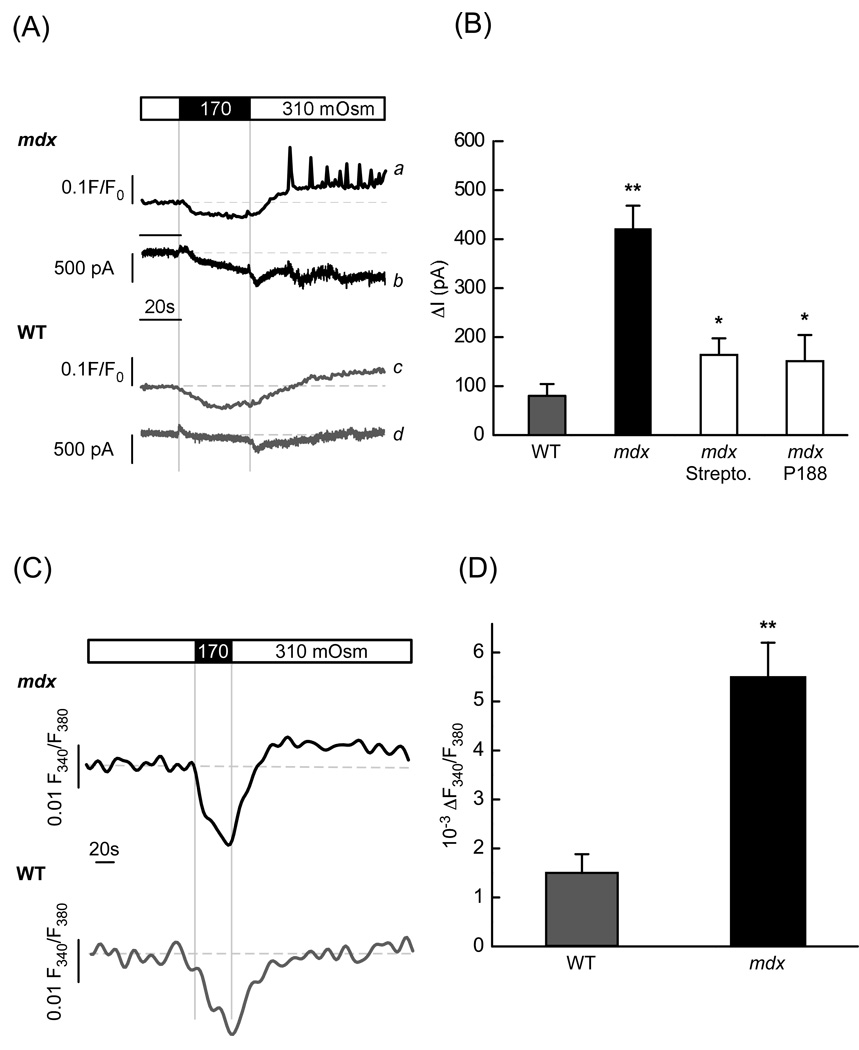
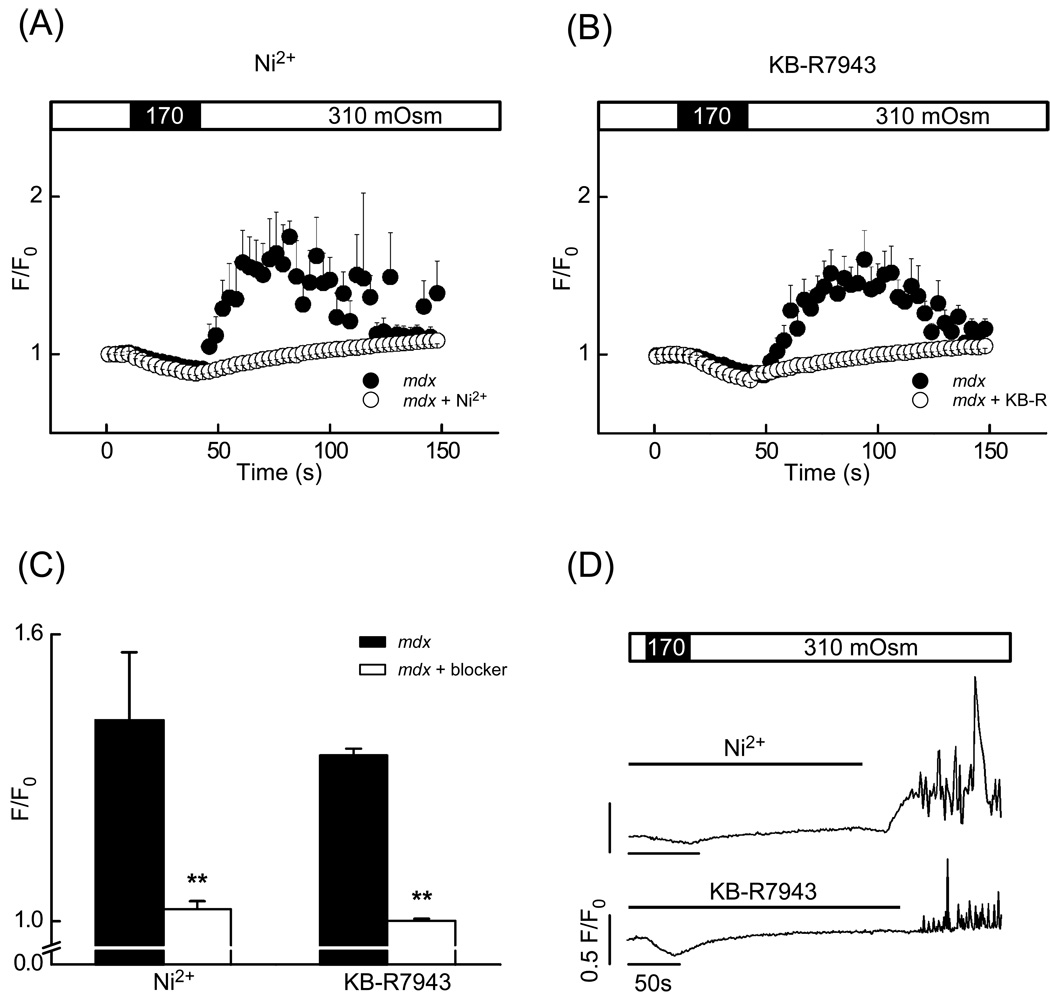
In Duchenne muscular dystrophy, deficiency of the cytoskeletal protein dystrophin leads to well-described defects in skeletal muscle, but also to dilated cardiomyopathy, accounting for about 20% of the mortality. Mechanisms leading to cardiomyocyte cell death and cardiomyopathy are not well understood. One hypothesis suggests that the lack of dystrophin leads to membrane instability during mechanical stress and to activation of Ca2+ entry pathways. Using cardiomyocytes isolated from dystrophic mdx mice we dissected the contribution of various putative Ca2+ influx pathways with pharmacological tools. Cytosolic Ca2+ and Na+ signals as well as uptake of membrane impermeant compounds were monitored with fluorescent indicators using confocal microscopy and photometry. Membrane stress was applied as moderate osmotic challenges while membrane current was quantified using the whole-cell patch-clamp technique. Our findings suggest a major contribution of two primary Ca2+ influx pathways, stretch-activated membrane channels and short-lived microruptures. Furthermore, we found evidence for a secondary Ca2+ influx pathway, the Na+-Ca2+ exchange (NCX), which in cardiac muscle has a large transport capacity. After stress it contributes to Ca2+ entry in exchange for Na+ which had previously entered via primary stress-induced pathways, representing a previously not recognized mechanism contributing to subsequent cellular damage. This complexity needs to be considered when targeting abnormal Ca2+ influx as a treatment option for dystrophy.
Figures
References
-
- Ervasti JM, Campbell KP. Dystrophin and the membrane skeleton. Curr Opin Cell Biol. 1993;5:82–87. - PubMed
-
- Finsterer J, Stollberger C. The heart in human dystrophinopathies. Cardiology. 2003;99:1–19. - PubMed
-
- Quinlan JG, Hahn HS, Wong BL, Lorenz JN, Wenisch AS, Levin LS. Evolution of the mdx mouse cardiomyopathy: physiological and morphological findings. Neuromuscul Disord. 2004;14:491–496. - PubMed
-
- Jung C, Martins AS, Niggli E, Shirokova N. Dystrophic cardiomyopathy: amplification of cellular damage by Ca2+ signalling and reactive oxygen species-generating pathways. Cardiovasc Res. 2008;77:766–773. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
