

Shotgun proteomics in neuroscience
- PMID: 19607789
- PMCID: PMC2746002
- DOI: 10.1016/j.neuron.2009.06.011
Shotgun proteomics in neuroscience
Abstract
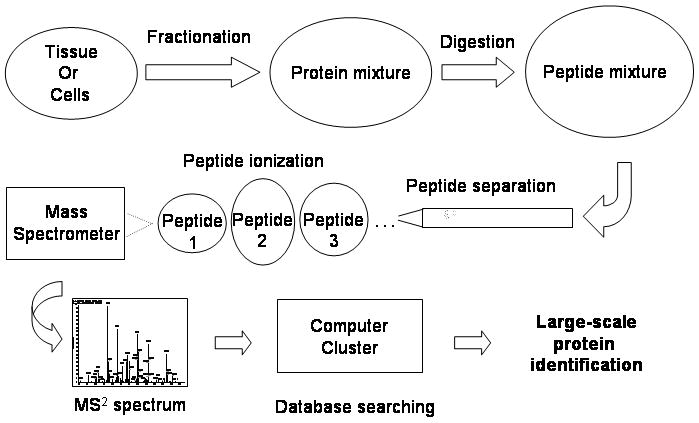
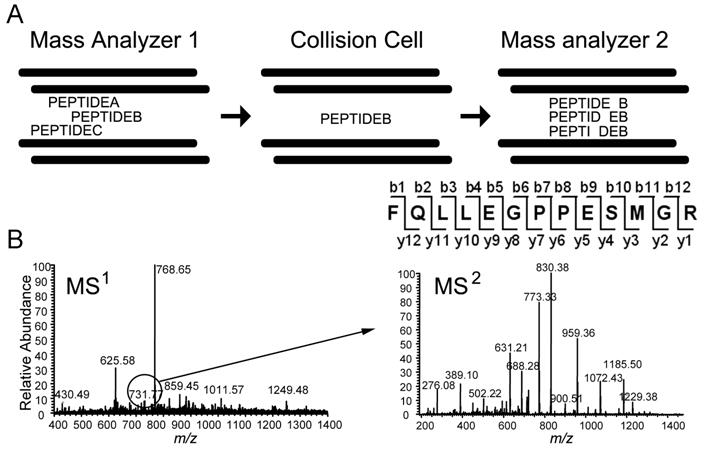
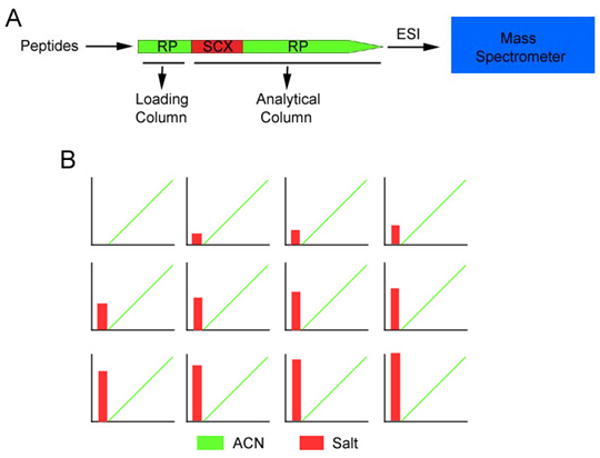
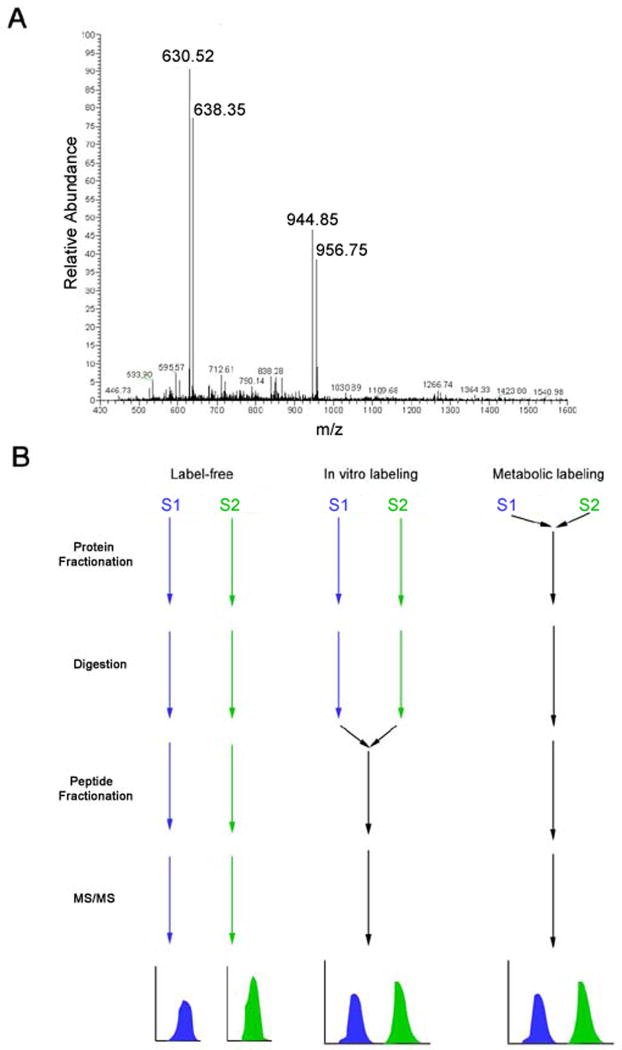
Mass spectrometry-based proteomics is increasingly used to address basic and clinical questions in biomedical research through studies of differential protein expression, protein-protein interactions, and posttranslational modifications. The complex structural and functional organization of the human brain warrants the application of high-throughput, systematic approaches to understand the functional alterations under normal physiological conditions and the perturbations of neurological diseases. This primer focuses on shotgun-proteomics-based tandem mass spectrometry for the identification of proteins in a complex mixture. It describes the basic concepts of protein differential expression analysis and posttranslational modification analysis and discusses several strategies to improve the coverage of the proteome.
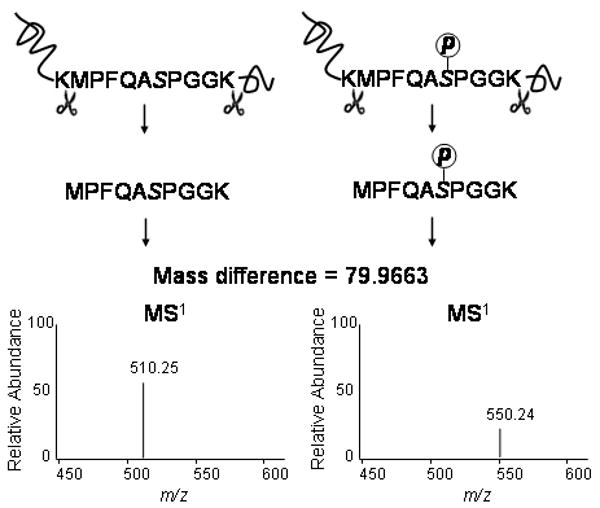
Figures
Similar articles
-
Approaches for systematic proteome exploration.Biomol Eng. 2007 Jun;24(2):155-68. doi: 10.1016/j.bioeng.2007.01.001. Epub 2007 Feb 6. Biomol Eng. 2007. PMID: 17376740 Review.
-
A Primer on Concepts and Applications of Proteomics in Neuroscience.Neuron. 2017 Nov 1;96(3):558-571. doi: 10.1016/j.neuron.2017.09.025. Neuron. 2017. PMID: 29096073 Review.
-
Regulation of developmental processes: insights from mass spectrometry-based proteomics.Wiley Interdiscip Rev Dev Biol. 2013 Sep-Oct;2(5):723-34. doi: 10.1002/wdev.102. Epub 2012 Dec 6. Wiley Interdiscip Rev Dev Biol. 2013. PMID: 24014456 Free PMC article. Review.
-
To label or not to label: applications of quantitative proteomics in neuroscience research.Proteomics. 2012 Feb;12(4-5):736-47. doi: 10.1002/pmic.201100350. Epub 2012 Jan 23. Proteomics. 2012. PMID: 22247077 Review.
-
Functional proteomics: mapping protein-protein interactions and pathways.Curr Opin Mol Ther. 2002 Jun;4(3):210-5. Curr Opin Mol Ther. 2002. PMID: 12139305 Review.
Cited by
-
Comparative analyses of nuclear proteome: extending its function.Front Plant Sci. 2013 Apr 26;4:100. doi: 10.3389/fpls.2013.00100. eCollection 2013. Front Plant Sci. 2013. PMID: 23637696 Free PMC article.
-
Network organization of the huntingtin proteomic interactome in mammalian brain.Neuron. 2012 Jul 12;75(1):41-57. doi: 10.1016/j.neuron.2012.05.024. Neuron. 2012. PMID: 22794259 Free PMC article.
-
Pulsed Azidohomoalanine Labeling in Mammals (PALM) Detects Changes in Liver-Specific LKB1 Knockout Mice.J Proteome Res. 2015 Nov 6;14(11):4815-22. doi: 10.1021/acs.jproteome.5b00653. Epub 2015 Oct 26. J Proteome Res. 2015. PMID: 26445171 Free PMC article.
-
Identification of functional marker proteins in the mammalian growth cone.Proc Natl Acad Sci U S A. 2009 Oct 6;106(40):17211-6. doi: 10.1073/pnas.0904092106. Epub 2009 Sep 28. Proc Natl Acad Sci U S A. 2009. PMID: 19805073 Free PMC article.
-
Discovery of Novel Cell Surface Markers for Purification of Embryonic Dopamine Progenitors for Transplantation in Parkinson's Disease Animal Models.Mol Cell Proteomics. 2018 Sep;17(9):1670-1684. doi: 10.1074/mcp.RA118.000809. Epub 2018 May 29. Mol Cell Proteomics. 2018. PMID: 29848781 Free PMC article.
References
-
- Aguilar MI, Hearn MT. High-resolution reversed-phase high-performance liquid chromatography of peptides and proteins. Methods Enzymol. 1996;270:3–26. - PubMed
-
- America AH, Cordewener JH. Comparative LC-MS: a landscape of peaks and valleys. Proteomics. 2008;8:731–749. - PubMed
-
- Anderle M, Roy S, Lin H, Becker C, Joho K. Quantifying reproducibility for differential proteomics: noise analysis for protein liquid chromatography-mass spectrometry of human serum. Bioinformatics. 2004;20:3575–3582. - PubMed
-
- Anderson L, Hunter CL. Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol Cell Proteomics. 2006;5:573–588. - PubMed
-
- Andersson L, Porath J. Isolation of phosphoproteins by immobilized metal (Fe3+) affinity chromatography. Anal Biochem. 1986;154:250–254. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
