

Redox modification of ryanodine receptors underlies calcium alternans in a canine model of sudden cardiac death
- PMID: 19617226
- PMCID: PMC2777950
- DOI: 10.1093/cvr/cvp246
Redox modification of ryanodine receptors underlies calcium alternans in a canine model of sudden cardiac death
Abstract
Aims: Although cardiac alternans is a known predictor of lethal arrhythmias, its underlying causes remain largely undefined in disease settings. The potential role of, and mechanisms responsible for, beat-to-beat alternations in the amplitude of systolic Ca(2+) transients (Ca(2+) alternans) was investigated in a canine post-myocardial infarction (MI) model of sudden cardiac death (SCD).
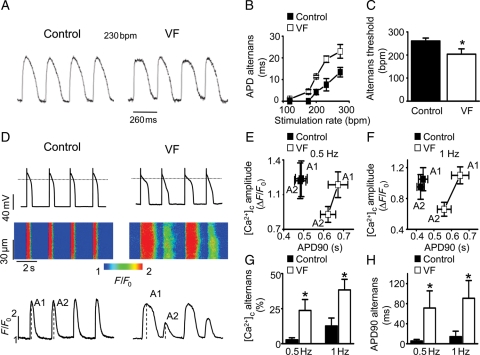
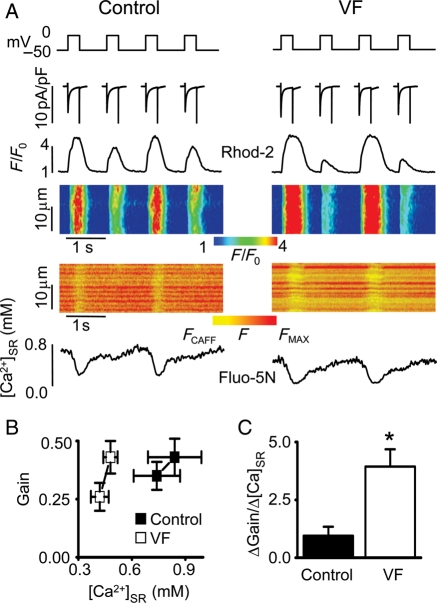
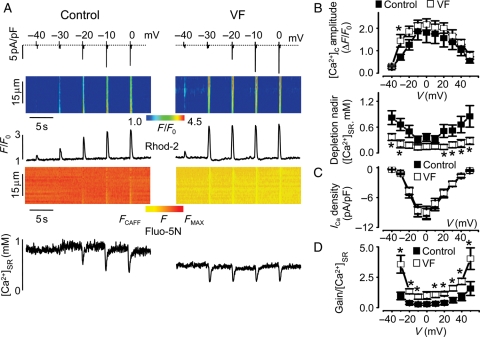
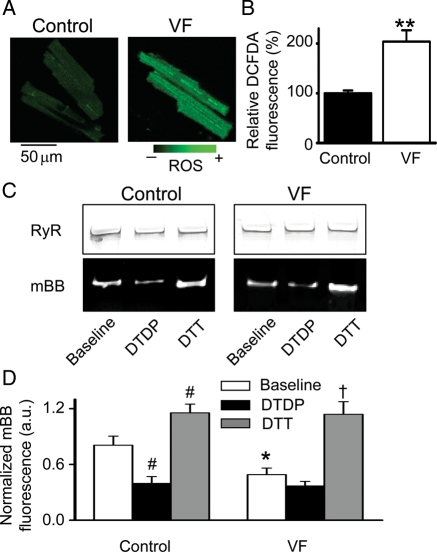
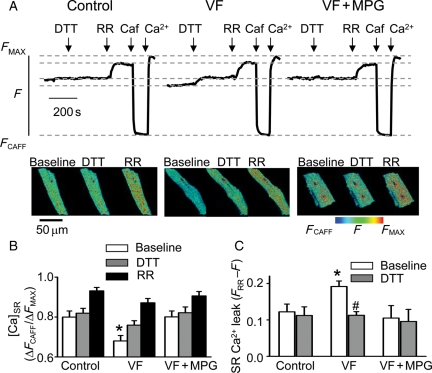
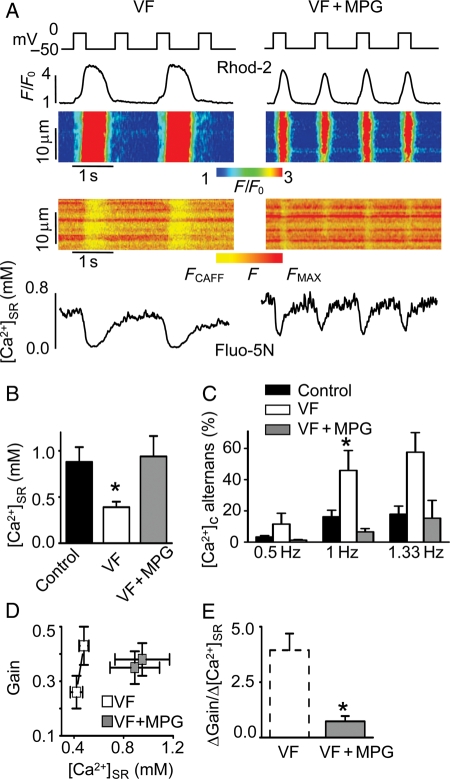
Methods and results: Post-MI dogs had preserved left ventricular (LV) function and susceptibility to ventricular fibrillation (VF) during exercise. LV wedge preparations from VF dogs were more susceptible to action potential (AP) alternans and the frequency-dependence of Ca(2+) alternans was shifted towards slower rates in myocytes isolated from VF dogs relative to controls. In both groups of cells, cytosolic Ca(2+) transients ([Ca(2+)](c)) alternated in phase with changes in diastolic Ca(2+) in sarcoplasmic reticulum ([Ca(2+)](SR)), but the dependence of [Ca(2+)](c) amplitude on [Ca(2+)](SR) was steeper in VF cells. Abnormal ryanodine receptor (RyR) function in VF cells was indicated by increased fractional Ca(2+) release for a given amplitude of Ca(2+) current and elevated diastolic RyR-mediated SR Ca(2+) leak. SR Ca(2+) uptake activity did not differ between VF and control cells. VF myocytes had an increased rate of reactive oxygen species production and increased RyR oxidation. Treatment of VF myocytes with reducing agents normalized parameters of Ca(2+) handling and shifted the threshold of Ca(2+) alternans to higher frequencies.
Conclusion: Redox modulation of RyRs promotes generation of Ca(2+) alternans by enhancing the steepness of the Ca(2+) release-load relationship and thereby providing a substrate for post-MI arrhythmias.
Figures
Comment in
-
'Leaky' ryanodine receptors and sudden cardiac death.Cardiovasc Res. 2009 Dec 1;84(3):343-4. doi: 10.1093/cvr/cvp330. Epub 2009 Oct 8. Cardiovasc Res. 2009. PMID: 19815562 No abstract available.
References
-
- Moss AJ, Zareba W, Hall WJ, Klein H, Wilber DJ, Cannom DS, et al. Prophylactic implantation of a defibrillator in patients with myocardial infarction and reduced ejection fraction. N Engl J Med. 2002;346:877–883. - PubMed
-
- Bunch TJ, Hohnloser SH, Gersh BJ. Mechanisms of sudden cardiac death in myocardial infarction survivors: insights from the randomized trials of implantable cardioverter-defibrillators. Circulation. 2007;115:2451–2457. - PubMed
-
- Clements-Jewery H, Andrag E, Curtis MJ. Druggable targets for sudden cardiac death prevention: lessons from the past and strategies for the future. Curr Opin Pharmacol. 2009;9:146–153. - PubMed
-
- Janse MJ, Wit AL. Electrophysiological mechanisms of ventricular arrhythmias resulting from myocardial ischemia and infarction. Physiol Rev. 1989;69:1049–1169. - PubMed
-
- Walker ML, Rosenbaum DS. Repolarization alternans: implications for the mechanism and prevention of sudden cardiac death. Cardiovasc Res. 2003;57:599–614. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
