

Changes in thermodynamic stability of von Willebrand factor differentially affect the force-dependent binding to platelet GPIbalpha
- PMID: 19619477
- PMCID: PMC2711320
- DOI: 10.1016/j.bpj.2009.05.009
Changes in thermodynamic stability of von Willebrand factor differentially affect the force-dependent binding to platelet GPIbalpha
Abstract
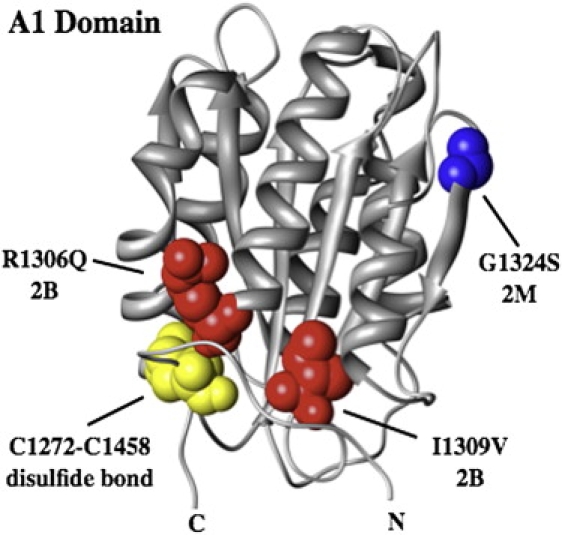
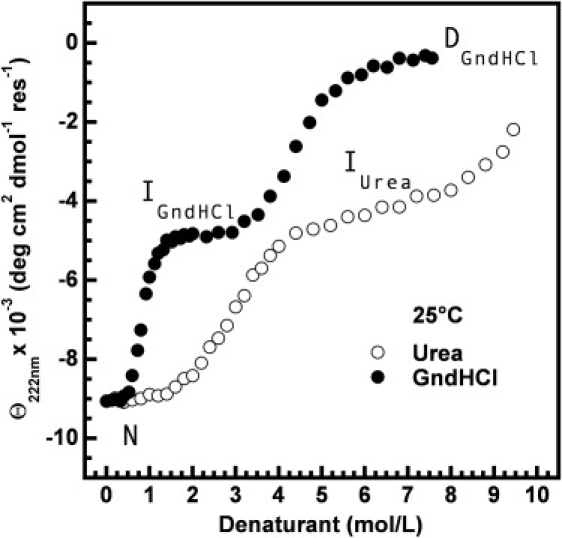
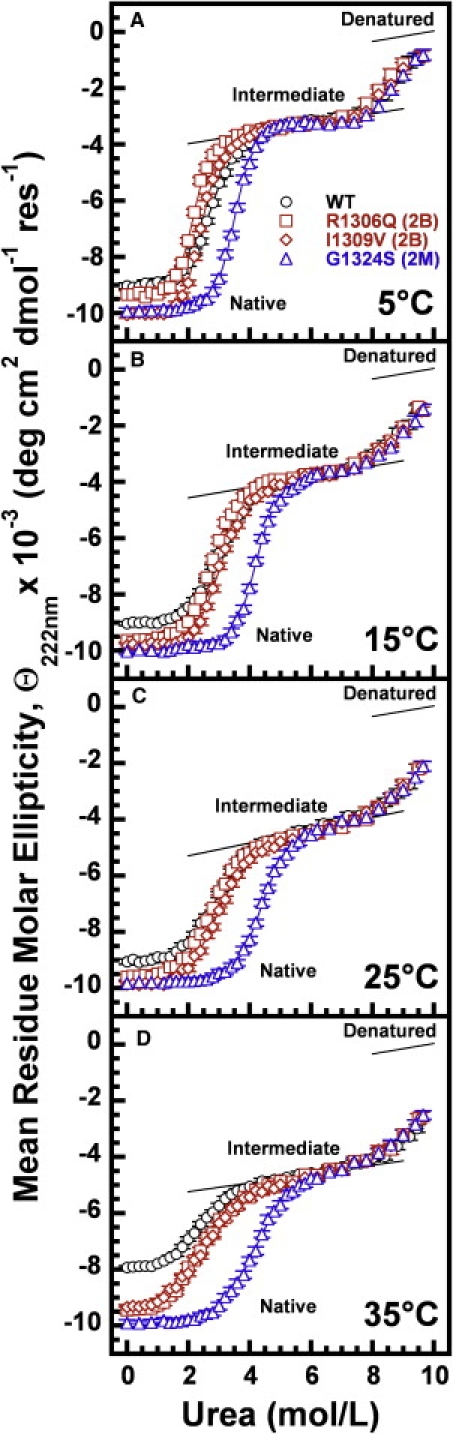
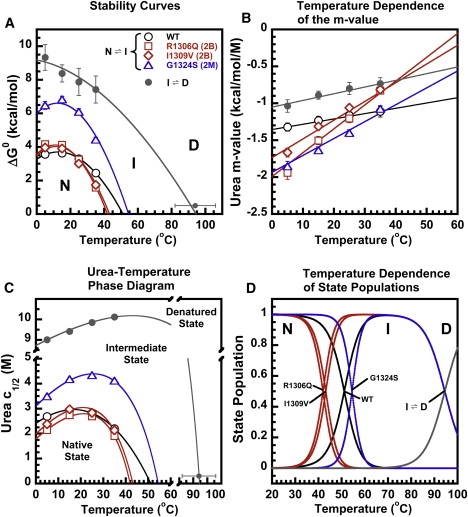
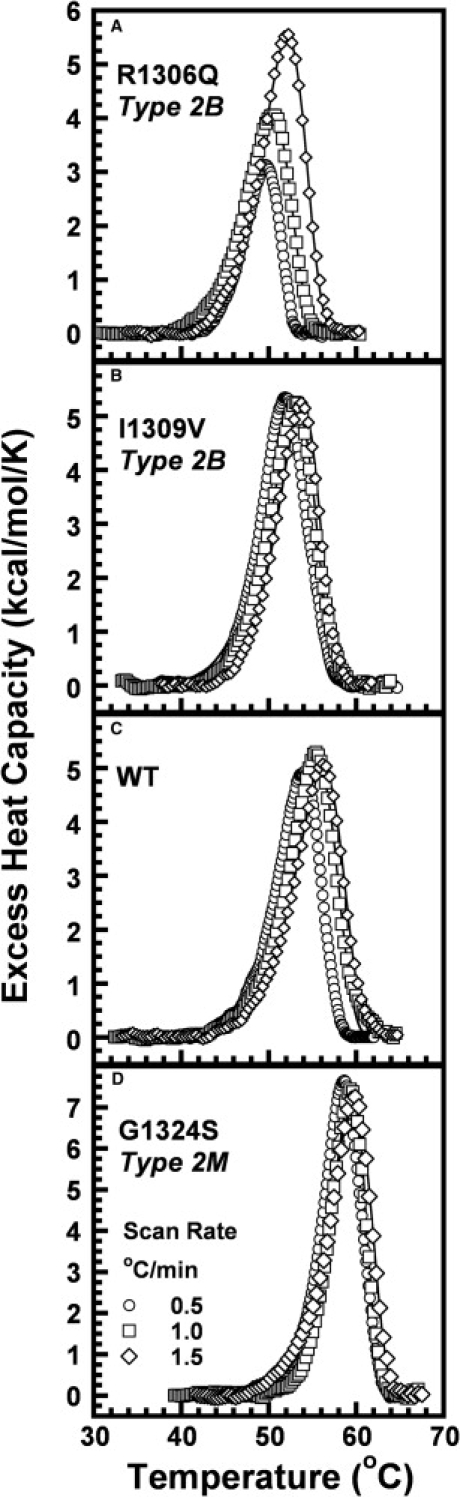
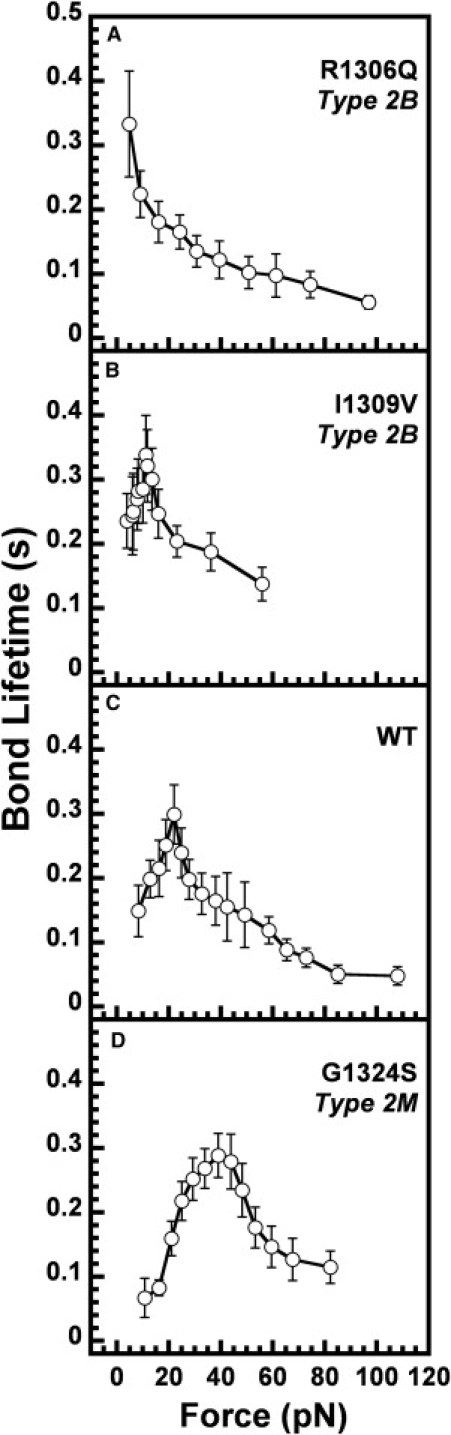
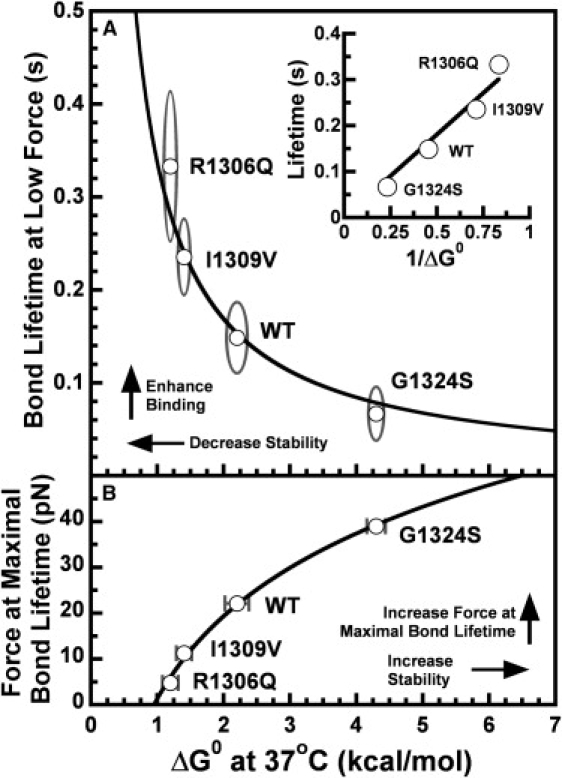
In circulation, plasma glycoprotein von Willebrand Factor plays an important role in hemostasis and in pathological thrombosis under hydrodynamic forces. Mutations in the A1 domain of von Willebrand factor cause the hereditary types 2B and 2M von Willebrand disease that either enhance (2B) or inhibit (2M) the interaction of von Willebrand factor with the platelet receptor glycoprotein Ibalpha. To understand how type 2B and 2M mutations cause clinically opposite phenotypes, we use a combination of protein unfolding thermodynamics and atomic force microscopy to assess the effects of two type 2B mutations (R1306Q and I1309V) and a type 2M mutation (G1324S) on the conformational stability of the A1 domain and the single bond dissociation kinetics of the A1-GPIbalpha interaction. At physiological temperature, the type 2B mutations destabilize the structure of the A1 domain and shift the A1-GPIbalpha catch to slip bonding to lower forces. Conversely, the type 2M mutation stabilizes the structure of the A1 domain and shifts the A1-GPIbalpha catch to slip bonding to higher forces. As a function of increasing A1 domain stability, the bond lifetime at low force decreases and the critical force required for maximal bond lifetime increases. Our results are able to distinguish the clinical phenotypes of these naturally occurring mutations from a thermodynamic and biophysical perspective that provides a quantitative description of the allosteric coupling of A1 conformational stability with the force dependent catch to slip bonding between A1 and GPIbalpha.
Figures
References
-
- von Willebrand E. Hereditary pseudohemophilia. Finska Läkarssällskapetes Handl. 1926;68:87–112.
-
- Federici A.B., Mannucci P.M. Management of inherited von Willebrand disease in 2007. Ann. Med. 2007;39:346–358. - PubMed
-
- Keeney S., Cumming A.M. The molecular biology of von Willebrand disease. Clin. Lab. Haematol. 2001;23:209–230. - PubMed
-
- Sadler J.E. Biochemistry and genetics of von Willebrand factor. Annu. Rev. Biochem. 1998;67:395–424. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
