

Evolution of the Schlafen genes, a gene family associated with embryonic lethality, meiotic drive, immune processes and orthopoxvirus virulence
- PMID: 19619625
- PMCID: PMC9533870
- DOI: 10.1016/j.gene.2009.07.006
Evolution of the Schlafen genes, a gene family associated with embryonic lethality, meiotic drive, immune processes and orthopoxvirus virulence
Abstract
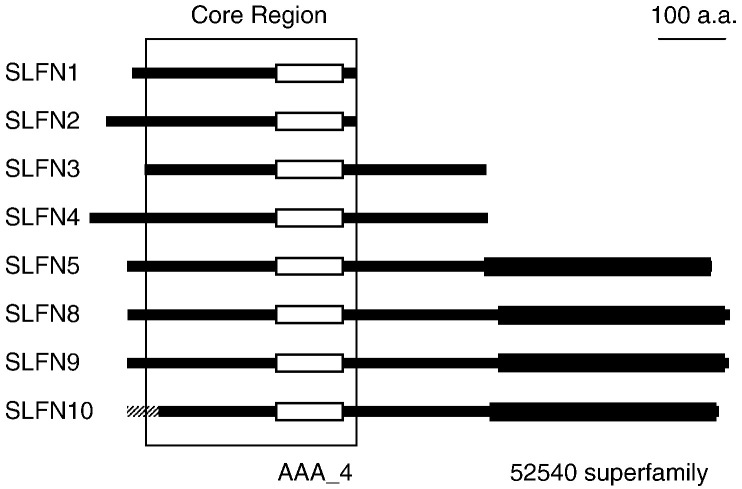
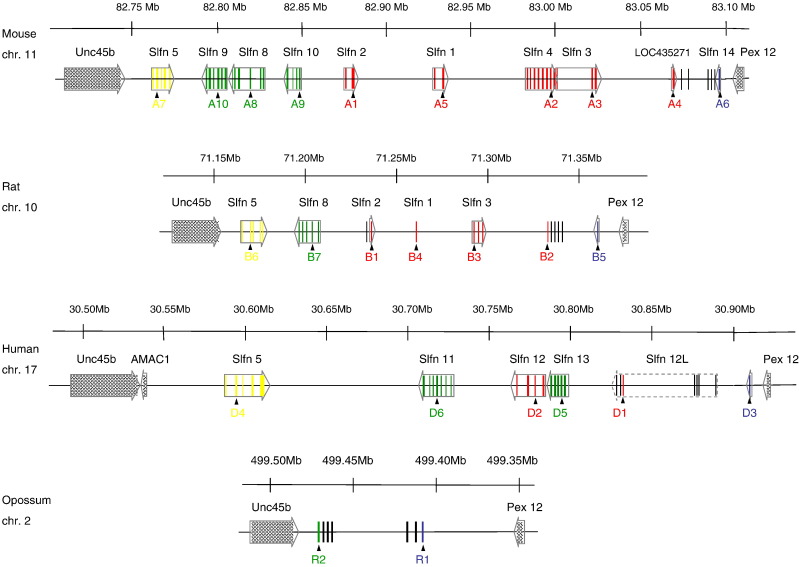
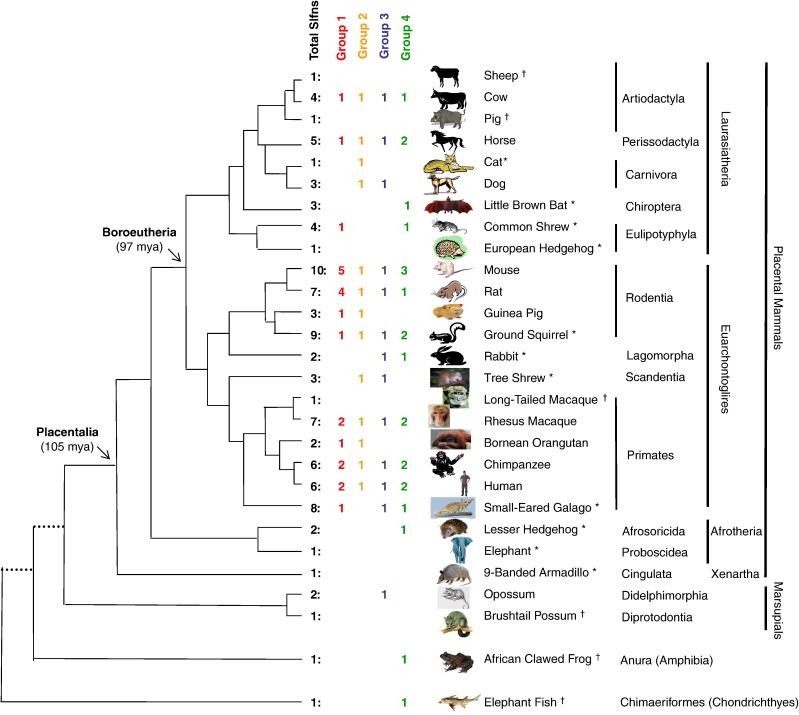
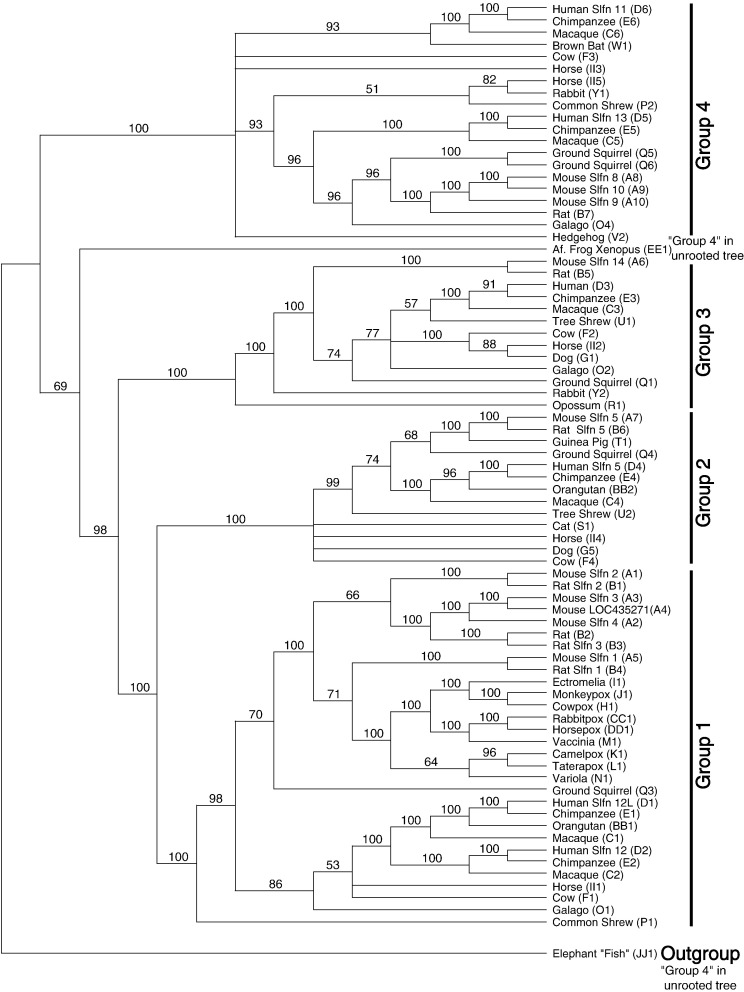
Genes of the Schlafen family, first discovered in mouse, are expressed in hematopoietic cells and are involved in immune processes. Previous results showed that they are candidate genes for two major phenomena: meiotic drive and embryonic lethality (DDK syndrome). However, these genes remain poorly understood, mostly due to the limitations imposed by their similarity, close location and the potential functional redundancy of the gene family members. Here we use genomic and phylogenetic studies to investigate the evolution and role of this family of genes. Our results show that the Schlafen family is widely distributed in mammals, where we recognize four major clades that experienced lineage-specific expansions or contractions in various orders, including primates and rodents. In addition, we identified members of the Schlafen family in Chondrichthyes and Amphibia, indicating an ancient origin of these genes. We find evidence that positive selection has acted on many Schlafen genes. Moreover, our analyses indicate that a member of the Schlafen family was horizontally transferred from murine rodents to orthopoxviruses, where it is hypothesized to play a role in allowing the virus to survive host immune defense mechanisms. The functional relevance of the viral Schlafen sequences is further underscored by our finding that they are evolving under purifying selection. This is of particular importance, since orthopoxviruses infect mammals and include variola, the causative agent of smallpox, and monkeypox, an emerging virus of great concern for human health.
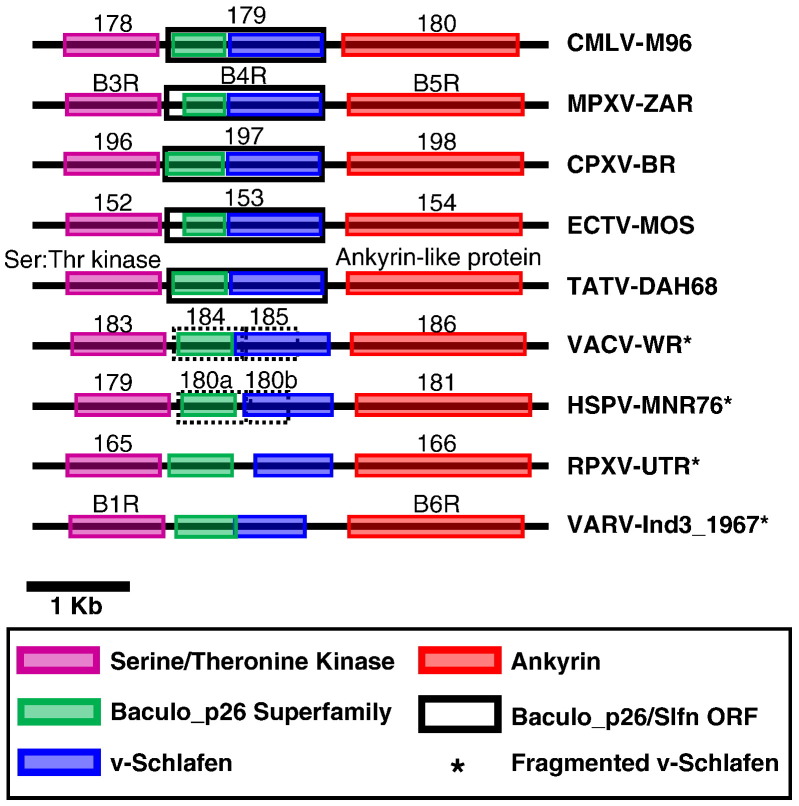
Figures

References
-
- Anisimova M., Bielawski J.P., Yang Z. Accuracy and power of the likelihood ratio test in detecting adaptive molecular evolution. Mol. Biol. Evol. 2001;18:1585–1592. - PubMed
-
- Anisimova M., Bielawski J.P., Yang Z. Accuracy and power of Bayes prediction of amino acid sites under positive selection. Mol. Biol. Evol. 2002;19:950–958. - PubMed
-
- Baldacci P.A., Richoux V., Renard J.P., Guénet J.L., Babinet C. The locus Om, responsible for the DDK syndrome, maps close to Sigje on mouse chromosome 11. Mamm. Genome. 1992;2:100–105. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
