

Multiple endocrine neoplasia type 1 knockout mice develop parathyroid, pancreatic, pituitary and adrenal tumours with hypercalcaemia, hypophosphataemia and hypercorticosteronaemia
- PMID: 19620250
- PMCID: PMC4439740
- DOI: 10.1677/ERC-09-0082
Multiple endocrine neoplasia type 1 knockout mice develop parathyroid, pancreatic, pituitary and adrenal tumours with hypercalcaemia, hypophosphataemia and hypercorticosteronaemia
Abstract
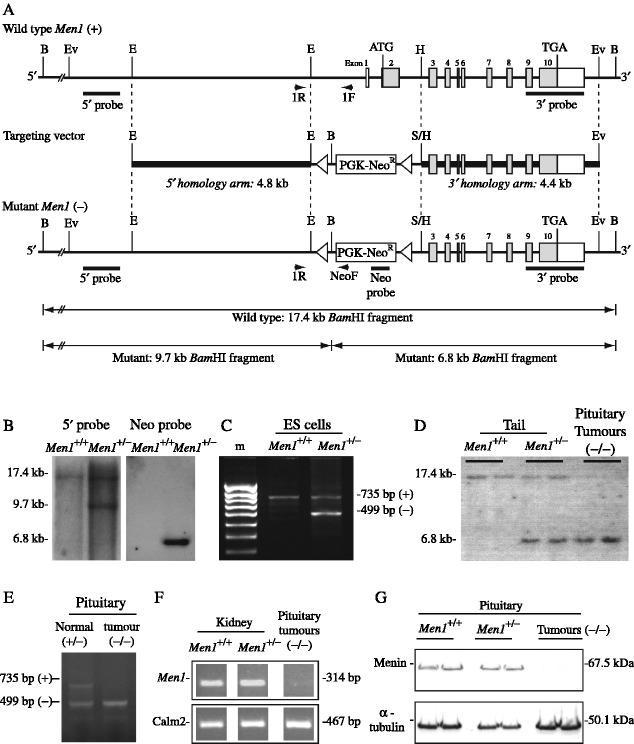
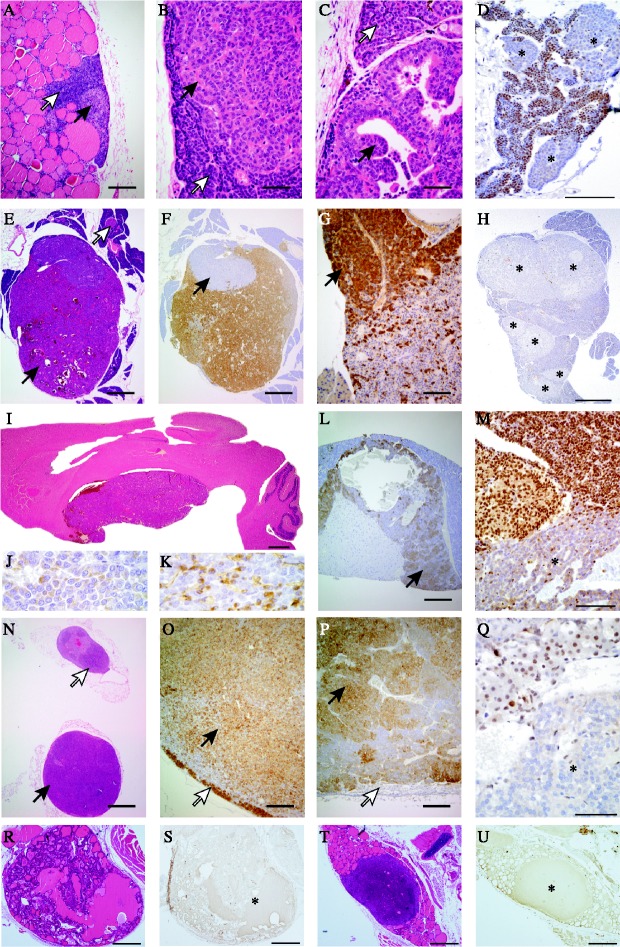
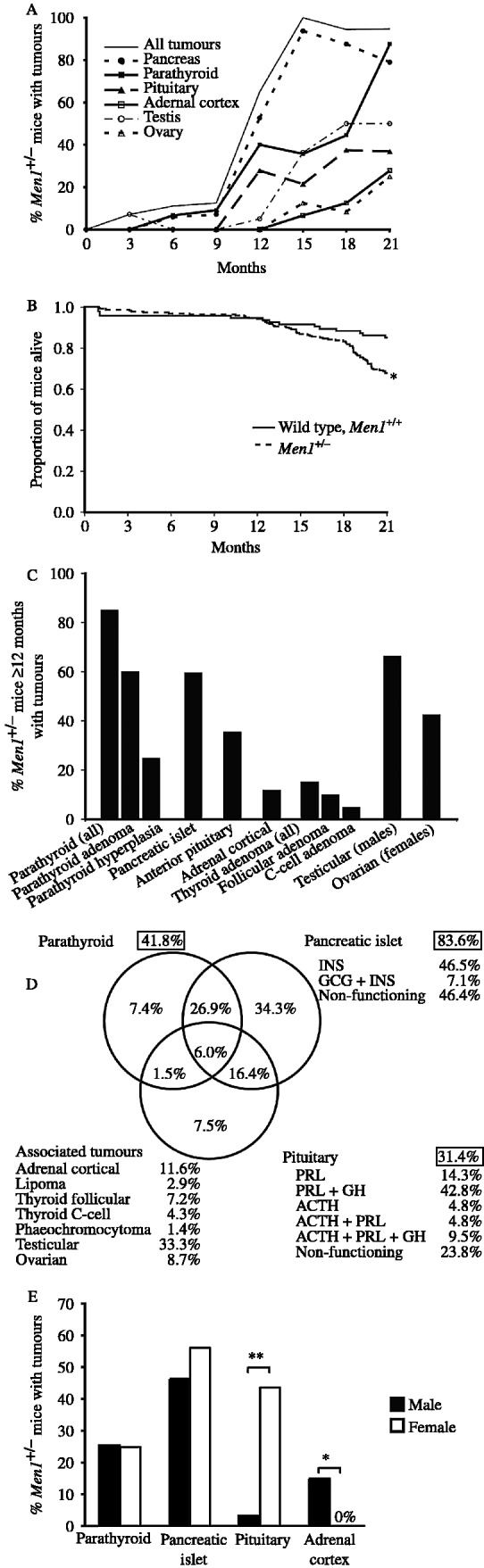
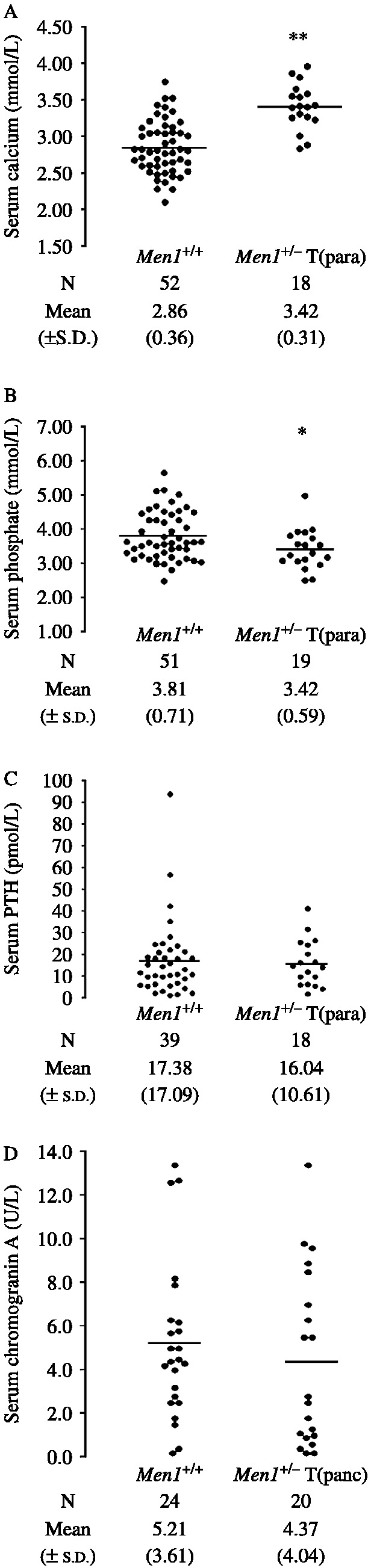
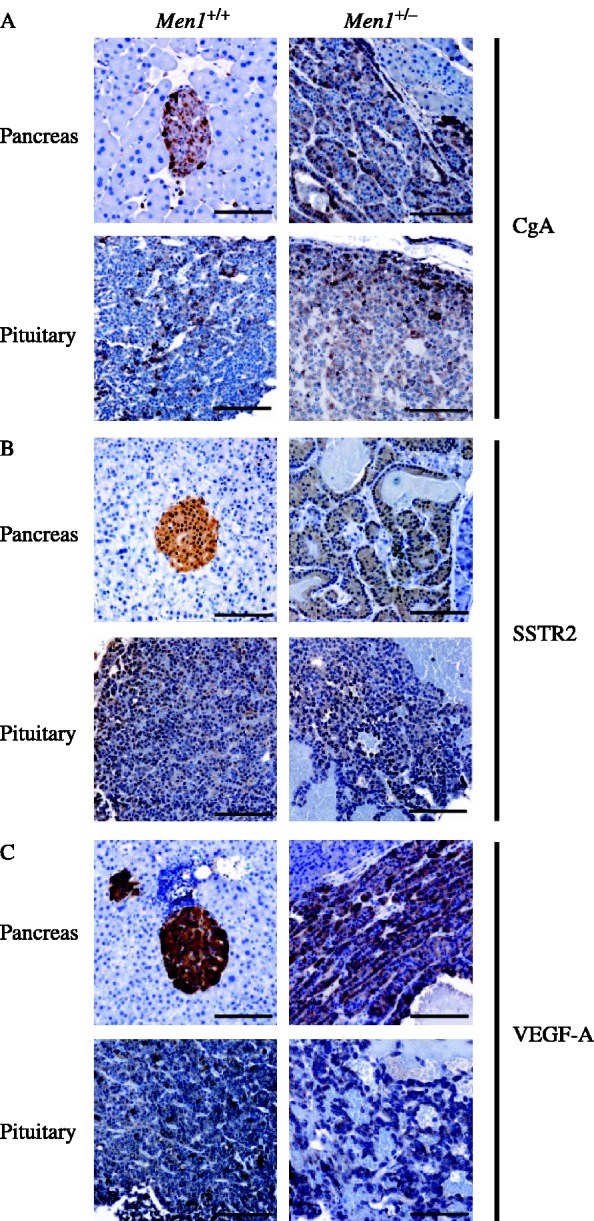
Multiple endocrine neoplasia type 1 (MEN1) is an autosomal dominant disorder characterized in man by parathyroid, pancreatic, pituitary and adrenal tumours. The MEN1 gene encodes a 610-amino acid protein (menin) which is a tumour suppressor. To investigate the in vivo role of menin, we developed a mouse model, by deleting Men1 exons 1 and 2 and investigated this for MEN1-associated tumours and serum abnormalities. Men1(+/-) mice were viable and fertile, and 220 Men1(+/-) and 94 Men1(+/+) mice were studied between the ages of 3 and 21 months. Survival in Men1(+/-) mice was significantly lower than in Men1(+/+) mice (<68% vs >85%, P<0.01). Men1(+/-) mice developed, by 9 months of age, parathyroid hyperplasia, pancreatic tumours which were mostly insulinomas, by 12 months of age, pituitary tumours which were mostly prolactinomas, and by 15 months parathyroid adenomas and adrenal cortical tumours. Loss of heterozygosity and menin expression was demonstrated in the tumours, consistent with a tumour suppressor role for the Men1 gene. Men1(+/-) mice with parathyroid neoplasms were hypercalcaemic and hypophosphataemic, with inappropriately normal serum parathyroid hormone concentrations. Pancreatic and pituitary tumours expressed chromogranin A (CgA), somatostatin receptor type 2 and vascular endothelial growth factor-A. Serum CgA concentrations in Men1(+/-) mice were not elevated. Adrenocortical tumours, which immunostained for 3-beta-hydroxysteroid dehydrogenase, developed in seven Men1(+/-) mice, but resulted in hypercorticosteronaemia in one out of the four mice that were investigated. Thus, these Men1(+/-) mice are representative of MEN1 in man, and will help in investigating molecular mechanisms and treatments for endocrine tumours.
Figures
Similar articles
-
Proliferation rates of multiple endocrine neoplasia type 1 (MEN1)-associated tumors.Endocrinology. 2012 Nov;153(11):5167-79. doi: 10.1210/en.2012-1675. Epub 2012 Sep 28. Endocrinology. 2012. PMID: 23024266 Free PMC article.
-
Multiple endocrine neoplasia type 1 (MEN1).Best Pract Res Clin Endocrinol Metab. 2010 Jun;24(3):355-70. doi: 10.1016/j.beem.2010.07.003. Best Pract Res Clin Endocrinol Metab. 2010. PMID: 20833329 Review.
-
Expression and functional analysis of menin in a multiple endocrine neoplasia type 1 (MEN1) patient with somatic loss of heterozygosity in chromosome 11q13 and unidentified germline mutation of the MEN1 gene.Endocrine. 2006 Jun;29(3):485-90. doi: 10.1385/ENDO:29:3:485. Endocrine. 2006. PMID: 16943588
-
Multiple endocrine neoplasia type 1.Orphanet J Rare Dis. 2006 Oct 2;1:38. doi: 10.1186/1750-1172-1-38. Orphanet J Rare Dis. 2006. PMID: 17014705 Free PMC article. Review.
-
A mouse model of multiple endocrine neoplasia, type 1, develops multiple endocrine tumors.Proc Natl Acad Sci U S A. 2001 Jan 30;98(3):1118-23. doi: 10.1073/pnas.98.3.1118. Proc Natl Acad Sci U S A. 2001. PMID: 11158604 Free PMC article.
Cited by
-
Genetic and epigenetic mutations of tumor suppressive genes in sporadic pituitary adenoma.Mol Cell Endocrinol. 2014 Apr 5;386(1-2):16-33. doi: 10.1016/j.mce.2013.09.006. Epub 2013 Sep 11. Mol Cell Endocrinol. 2014. PMID: 24035864 Free PMC article. Review.
-
Studies of mice deleted for Sox3 and uc482: relevance to X-linked hypoparathyroidism.Endocr Connect. 2020 Feb;9(2):173-186. doi: 10.1530/EC-19-0478. Endocr Connect. 2020. PMID: 31961795 Free PMC article.
-
A mouse model for inherited renal fibrosis associated with endoplasmic reticulum stress.Dis Model Mech. 2017 Jun 1;10(6):773-786. doi: 10.1242/dmm.029488. Epub 2017 Mar 21. Dis Model Mech. 2017. PMID: 28325753 Free PMC article.
-
siRNA Delivery for Control of Cyclin D1 and E2F1 Expression in Crohn's Disease.Transl Med UniSa. 2018 Mar 31;17:22-30. eCollection 2017 Jul. Transl Med UniSa. 2018. PMID: 30050877 Free PMC article.
-
Reversal of preexisting hyperglycemia in diabetic mice by acute deletion of the Men1 gene.Proc Natl Acad Sci U S A. 2010 Nov 23;107(47):20358-63. doi: 10.1073/pnas.1012257107. Epub 2010 Nov 8. Proc Natl Acad Sci U S A. 2010. PMID: 21059956 Free PMC article.
References
-
- Benson L, Ljunghall S, Akerstrom G, Oberg K. Hyperparathyroidism presenting as the first lesion in multiple endocrine neoplasia type 1. American Journal of Medicine. 1987;82:731–737. - PubMed
-
- Bertolino P, Tong WM, Galendo D, Wang ZQ, Zhang CX. Heterozygous Men1 mutant mice develop a range of endocrine tumors mimicking multiple endocrine neoplasia type 1. Molecular Endocrinology. 2003a;17:1880–1892. - PubMed
-
- Bertolino P, Tong WM, Herrera PL, Casse H, Zhang CX, Wang ZQ. Pancreatic beta-cell-specific ablation of the multiple endocrine neoplasia type 1 (MEN1) gene causes full penetrance of insulinoma development in mice. Cancer Research. 2003b;63:4836–4841. - PubMed
-
- Biondi CA, Gartside MG, Waring P, Loffler KA, Stark MS, Magnuson MA, Kay GF, Hayward NK. Conditional inactivation of the MEN1 gene leads to pancreatic and pituitary tumorigenesis but does not affect normal development of these tissues. Molecular and Cellular Biology. 2004;24:3125–3131. - PMC - PubMed
-
- Calender A, Giraud S, Cougard P, Chanson P, Lenoir G, Murat A, Hamon P, Proye C. Multiple endocrine neoplasia type 1 in France: clinical and genetic studies. Journal of Internal Medicine. 1995;238:263–268. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
