

Multiple endocrine neoplasia type 1 knockout mice develop parathyroid, pancreatic, pituitary and adrenal tumours with hypercalcaemia, hypophosphataemia and hypercorticosteronaemia
- PMID: 19620250
- PMCID: PMC4439740
- DOI: 10.1677/ERC-09-0082
Multiple endocrine neoplasia type 1 knockout mice develop parathyroid, pancreatic, pituitary and adrenal tumours with hypercalcaemia, hypophosphataemia and hypercorticosteronaemia
Abstract
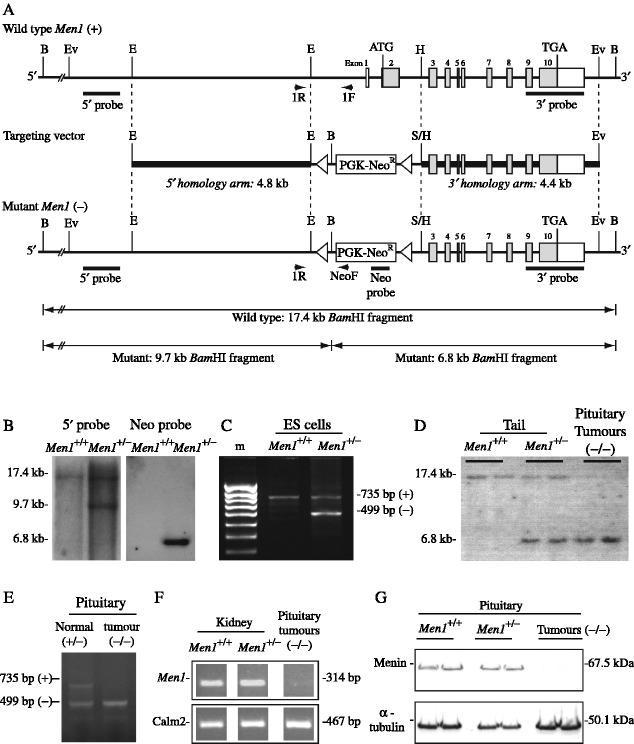
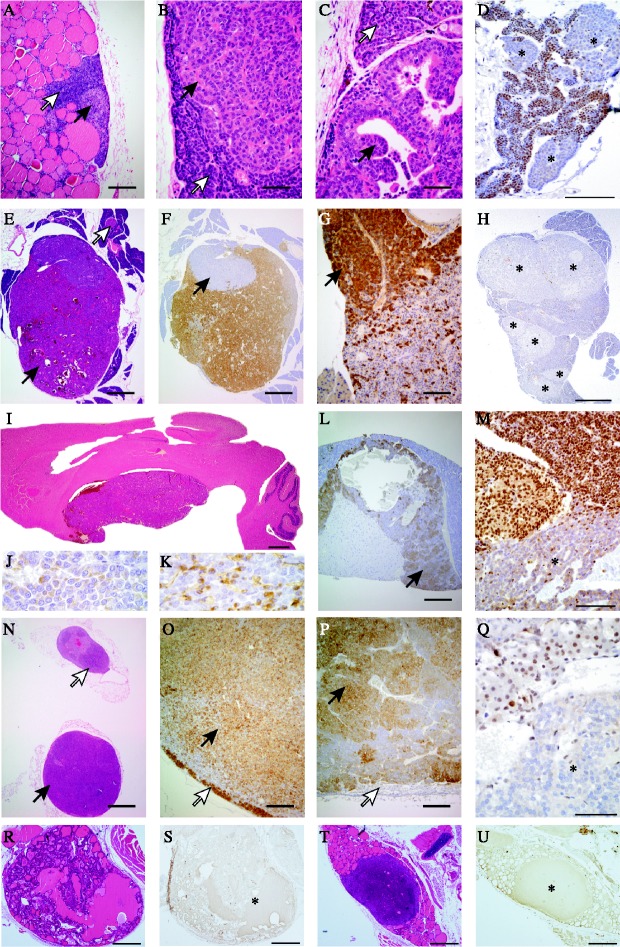
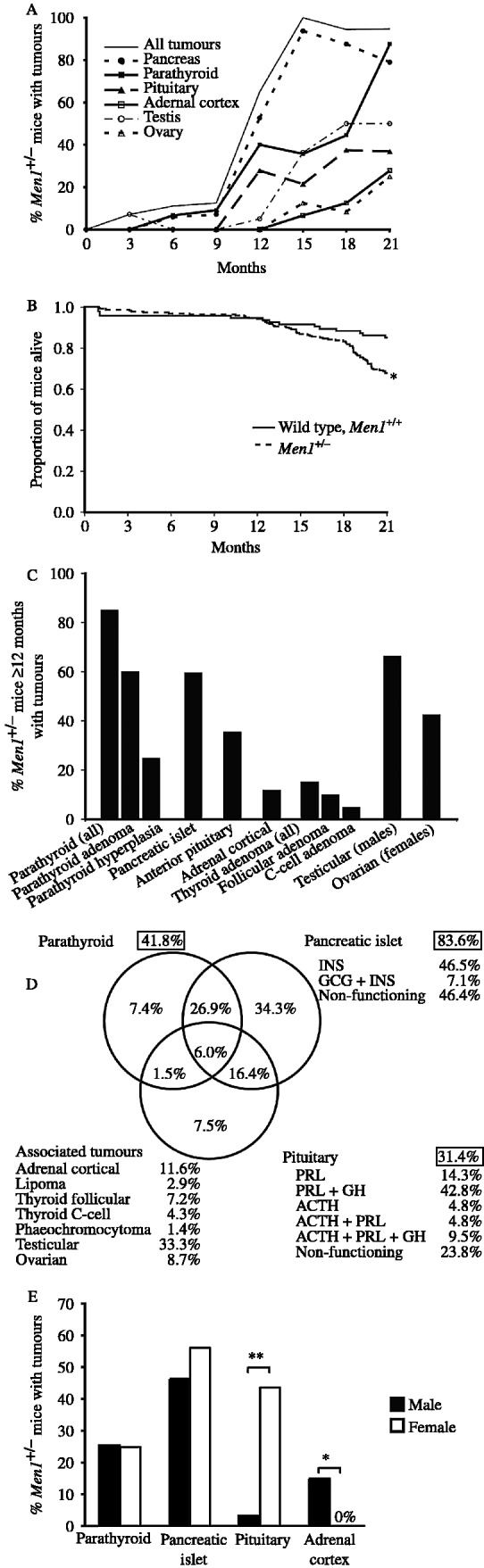
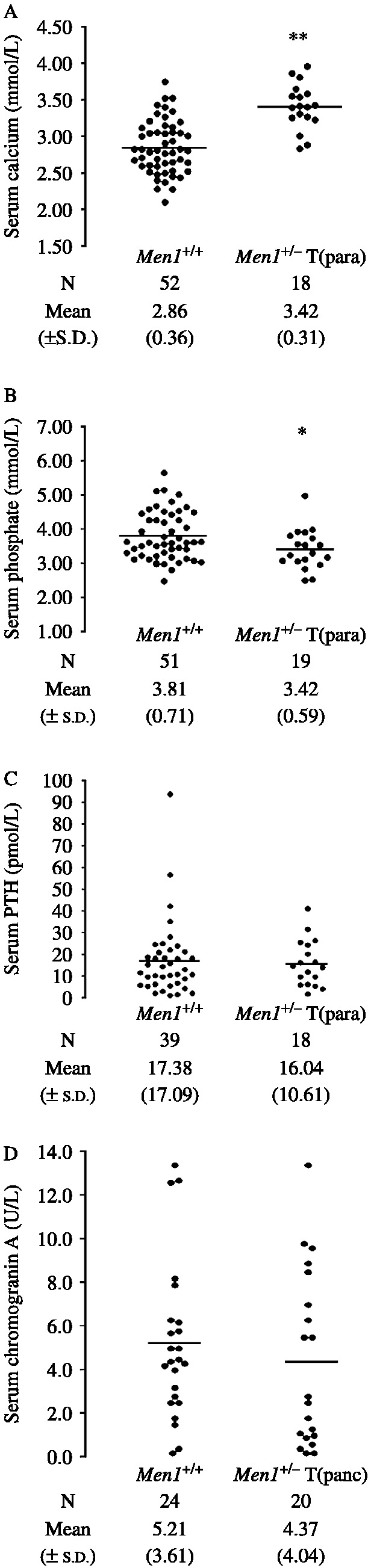
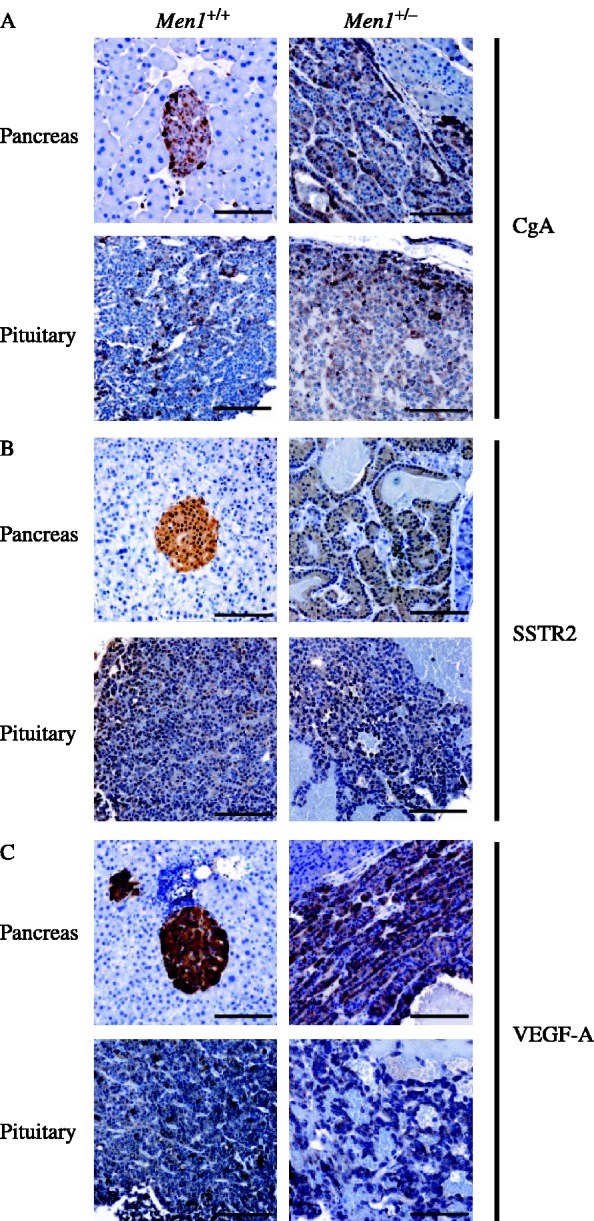
Multiple endocrine neoplasia type 1 (MEN1) is an autosomal dominant disorder characterized in man by parathyroid, pancreatic, pituitary and adrenal tumours. The MEN1 gene encodes a 610-amino acid protein (menin) which is a tumour suppressor. To investigate the in vivo role of menin, we developed a mouse model, by deleting Men1 exons 1 and 2 and investigated this for MEN1-associated tumours and serum abnormalities. Men1(+/-) mice were viable and fertile, and 220 Men1(+/-) and 94 Men1(+/+) mice were studied between the ages of 3 and 21 months. Survival in Men1(+/-) mice was significantly lower than in Men1(+/+) mice (<68% vs >85%, P<0.01). Men1(+/-) mice developed, by 9 months of age, parathyroid hyperplasia, pancreatic tumours which were mostly insulinomas, by 12 months of age, pituitary tumours which were mostly prolactinomas, and by 15 months parathyroid adenomas and adrenal cortical tumours. Loss of heterozygosity and menin expression was demonstrated in the tumours, consistent with a tumour suppressor role for the Men1 gene. Men1(+/-) mice with parathyroid neoplasms were hypercalcaemic and hypophosphataemic, with inappropriately normal serum parathyroid hormone concentrations. Pancreatic and pituitary tumours expressed chromogranin A (CgA), somatostatin receptor type 2 and vascular endothelial growth factor-A. Serum CgA concentrations in Men1(+/-) mice were not elevated. Adrenocortical tumours, which immunostained for 3-beta-hydroxysteroid dehydrogenase, developed in seven Men1(+/-) mice, but resulted in hypercorticosteronaemia in one out of the four mice that were investigated. Thus, these Men1(+/-) mice are representative of MEN1 in man, and will help in investigating molecular mechanisms and treatments for endocrine tumours.
Figures
References
-
- Benson L, Ljunghall S, Akerstrom G, Oberg K. Hyperparathyroidism presenting as the first lesion in multiple endocrine neoplasia type 1. American Journal of Medicine. 1987;82:731–737. - PubMed
-
- Bertolino P, Tong WM, Galendo D, Wang ZQ, Zhang CX. Heterozygous Men1 mutant mice develop a range of endocrine tumors mimicking multiple endocrine neoplasia type 1. Molecular Endocrinology. 2003a;17:1880–1892. - PubMed
-
- Bertolino P, Tong WM, Herrera PL, Casse H, Zhang CX, Wang ZQ. Pancreatic beta-cell-specific ablation of the multiple endocrine neoplasia type 1 (MEN1) gene causes full penetrance of insulinoma development in mice. Cancer Research. 2003b;63:4836–4841. - PubMed
-
- Biondi CA, Gartside MG, Waring P, Loffler KA, Stark MS, Magnuson MA, Kay GF, Hayward NK. Conditional inactivation of the MEN1 gene leads to pancreatic and pituitary tumorigenesis but does not affect normal development of these tissues. Molecular and Cellular Biology. 2004;24:3125–3131. - PMC - PubMed
-
- Calender A, Giraud S, Cougard P, Chanson P, Lenoir G, Murat A, Hamon P, Proye C. Multiple endocrine neoplasia type 1 in France: clinical and genetic studies. Journal of Internal Medicine. 1995;238:263–268. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
