

Pre-B cell receptor-mediated cell cycle arrest in Philadelphia chromosome-positive acute lymphoblastic leukemia requires IKAROS function
- PMID: 19620627
- PMCID: PMC2722172
- DOI: 10.1084/jem.20090004
Pre-B cell receptor-mediated cell cycle arrest in Philadelphia chromosome-positive acute lymphoblastic leukemia requires IKAROS function
Abstract
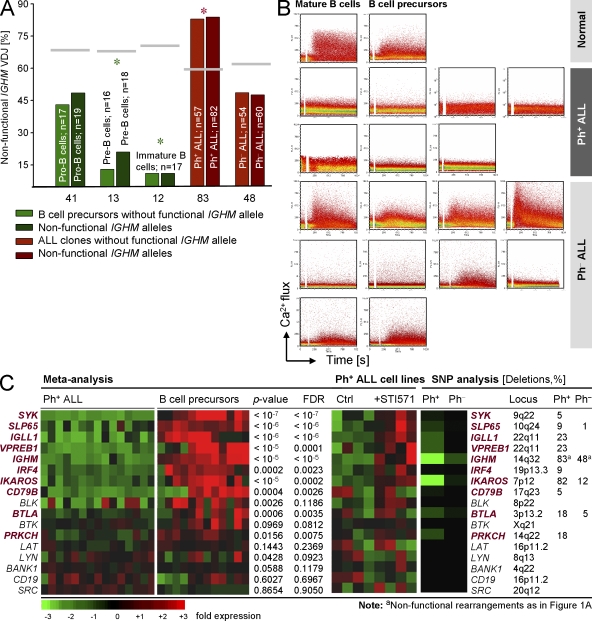
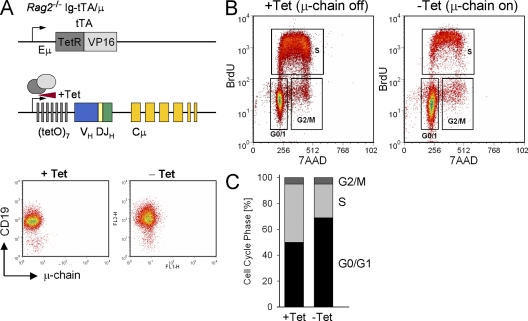
B cell lineage acute lymphoblastic leukemia (ALL) arises in virtually all cases from B cell precursors that are arrested at pre-B cell receptor-dependent stages. The Philadelphia chromosome-positive (Ph(+)) subtype of ALL accounts for 25-30% of cases of adult ALL, has the most unfavorable clinical outcome among all ALL subtypes and is defined by the oncogenic BCR-ABL1 kinase and deletions of the IKAROS gene in >80% of cases. Here, we demonstrate that the pre-B cell receptor functions as a tumor suppressor upstream of IKAROS through induction of cell cycle arrest in Ph(+) ALL cells. Pre-B cell receptor-mediated cell cycle arrest in Ph(+) ALL cells critically depends on IKAROS function, and is reversed by coexpression of the dominant-negative IKAROS splice variant IK6. IKAROS also promotes tumor suppression through cooperation with downstream molecules of the pre-B cell receptor signaling pathway, even if expression of the pre-B cell receptor itself is compromised. In this case, IKAROS redirects oncogenic BCR-ABL1 tyrosine kinase signaling from SRC kinase-activation to SLP65, which functions as a critical tumor suppressor downstream of the pre-B cell receptor. These findings provide a rationale for the surprisingly high frequency of IKAROS deletions in Ph(+) ALL and identify IKAROS-mediated cell cycle exit as the endpoint of an emerging pathway of pre-B cell receptor-mediated tumor suppression.
Figures





References
-
- Bäckesjö C.M., Vargas L., Superti-Furga G., Smith C.I. 2002. Phosphorylation of Bruton's tyrosine kinase by c-Abl.Biochem. Biophys. Res. Commun. 299:510–515 - PubMed
-
- Cheng A.M., Rowley B., Pao W., Hayday A., Bolen J.B., Pawson T. 1995. Syk tyrosine kinase required for mouse viability and B-cell development.Nature. 378:303–306 - PubMed
-
- Danial N.N., Pernis A., Rothman P.B. 1995. Jak-STAT signaling induced by the v-abl oncogene.Science. 269:1875–1877 - PubMed
-
- Flemming A., Brummer T., Reth M., Jumaa H. 2003. The adaptor protein SLP65 acts as a tumor suppressor that limits pre-B cell expansion.Nat. Immunol. 4:38–43 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
