

Impaired clearance of accumulated lysosomal glycogen in advanced Pompe disease despite high-level vector-mediated transgene expression
- PMID: 19621331
- PMCID: PMC3622249
- DOI: 10.1002/jgm.1372
Impaired clearance of accumulated lysosomal glycogen in advanced Pompe disease despite high-level vector-mediated transgene expression
Abstract
Background: Infantile-onset glycogen storage disease type II (GSD-II; Pompe disease; MIM 232300) causes death early in childhood from cardiorespiratory failure in the absence of effective treatment, whereas late-onset Pompe disease causes a progressive skeletal myopathy. The limitations of enzyme replacement therapy could potentially be addressed with adeno-associated virus (AAV) vector-mediated gene therapy.
Methods: AAV vectors containing tissue-specific regulatory cassettes, either liver-specific or muscle-specific, were administered to 12- and 17-month-old Pompe disease mice to evaluate the efficacy of gene therapy in advanced Pompe disease. Biochemical correction was evaluated through acid alpha-glucosidase (GAA) activity and glycogen content analyses of the heart and skeletal muscle. Western blotting, urinary biomarker, and Rotarod performance were evaluated after vector administration.
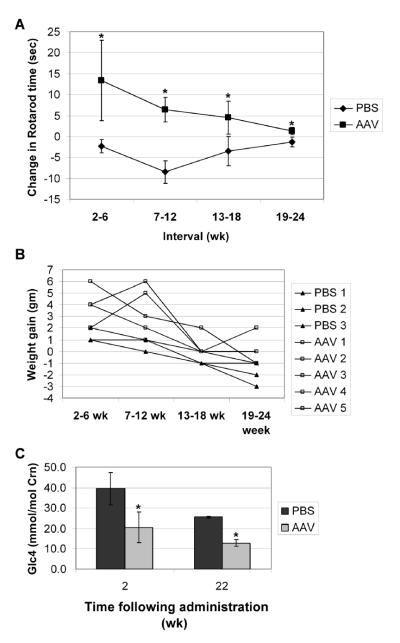
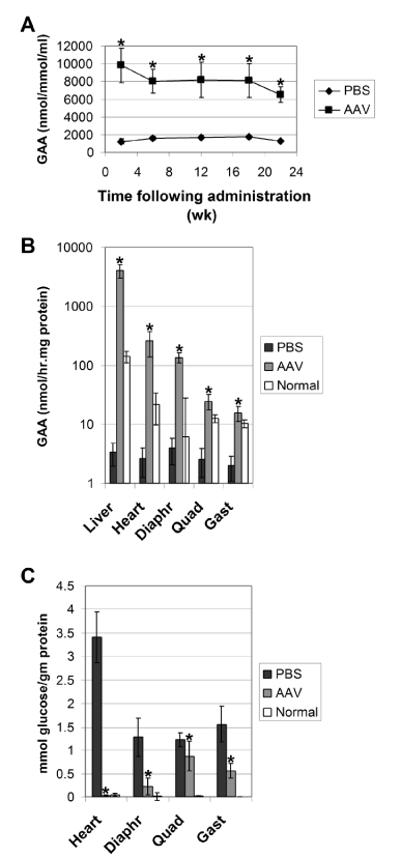
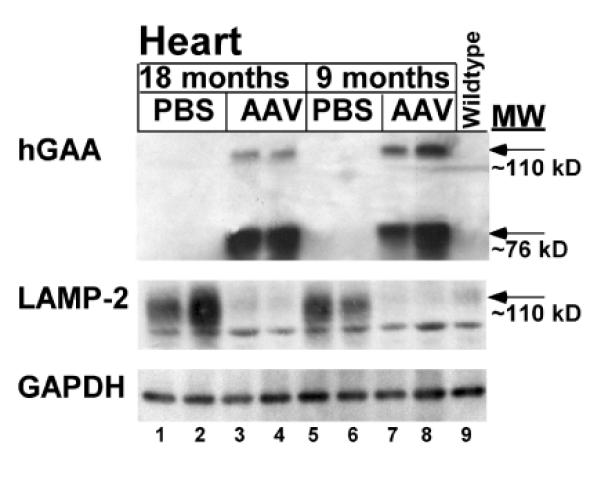
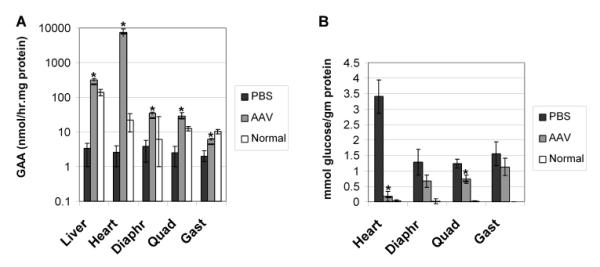
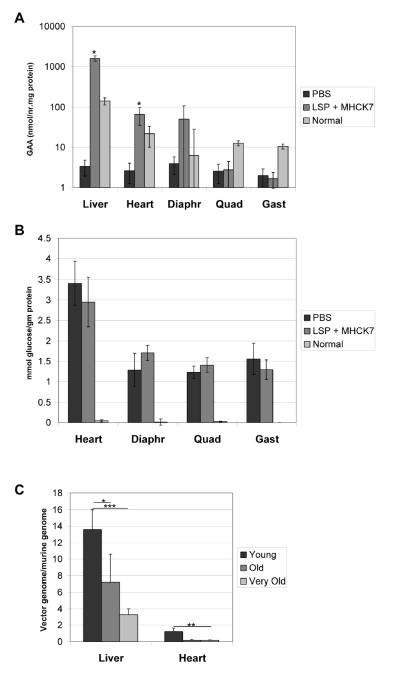
Results: The AAV vector containing the liver-specific regulatory cassette secreted high-level human GAA into the blood and corrected glycogen storage in the heart and diaphragm. The biochemical correction of the heart and diaphragm was associated with efficacy, as reflected by increased Rotarod performance; however, the clearance of glycogen from skeletal muscles was relatively impaired compared to in younger Pompe disease mice. An alternative vector containing a muscle-specific regulatory cassette transduced skeletal muscle with high efficiency, but also failed to achieve complete clearance of accumulated glycogen. Decreased transduction of the heart and liver in older mice, especially in females, was implicated as a cause for reduced efficacy in advanced Pompe disease.
Conclusions: The impaired efficacy of AAV vector-mediated gene therapy in old Pompe disease mice emphasizes the need for early treatment to achieve full efficacy.
Figures
Similar articles
-
Rescue of Advanced Pompe Disease in Mice with Hepatic Expression of Secretable Acid α-Glucosidase.Mol Ther. 2020 Sep 2;28(9):2056-2072. doi: 10.1016/j.ymthe.2020.05.025. Epub 2020 May 30. Mol Ther. 2020. PMID: 32526204 Free PMC article.
-
Antibody formation and mannose-6-phosphate receptor expression impact the efficacy of muscle-specific transgene expression in murine Pompe disease.J Gene Med. 2010 Nov;12(11):881-91. doi: 10.1002/jgm.1511. Epub 2010 Oct 22. J Gene Med. 2010. PMID: 20967919 Free PMC article.
-
Adjunctive β2-agonist treatment reduces glycogen independently of receptor-mediated acid α-glucosidase uptake in the limb muscles of mice with Pompe disease.FASEB J. 2014 May;28(5):2272-80. doi: 10.1096/fj.13-244202. Epub 2014 Jan 21. FASEB J. 2014. PMID: 24448824 Free PMC article.
-
Gene Therapy for Pompe Disease: The Time is now.Hum Gene Ther. 2019 Oct;30(10):1245-1262. doi: 10.1089/hum.2019.109. Epub 2019 Sep 9. Hum Gene Ther. 2019. PMID: 31298581 Review.
-
Immunomodulatory, liver depot gene therapy for Pompe disease.Cell Immunol. 2019 Aug;342:103737. doi: 10.1016/j.cellimm.2017.12.011. Epub 2017 Dec 29. Cell Immunol. 2019. PMID: 29295737 Free PMC article. Review.
Cited by
-
Rescue of Advanced Pompe Disease in Mice with Hepatic Expression of Secretable Acid α-Glucosidase.Mol Ther. 2020 Sep 2;28(9):2056-2072. doi: 10.1016/j.ymthe.2020.05.025. Epub 2020 May 30. Mol Ther. 2020. PMID: 32526204 Free PMC article.
-
Advances in Immune Tolerance Induction in Enzyme Replacement Therapy.Paediatr Drugs. 2024 May;26(3):287-308. doi: 10.1007/s40272-024-00627-9. Epub 2024 Apr 25. Paediatr Drugs. 2024. PMID: 38664313 Free PMC article. Review.
-
Antibody formation and mannose-6-phosphate receptor expression impact the efficacy of muscle-specific transgene expression in murine Pompe disease.J Gene Med. 2010 Nov;12(11):881-91. doi: 10.1002/jgm.1511. Epub 2010 Oct 22. J Gene Med. 2010. PMID: 20967919 Free PMC article.
-
Antibody-mediated enzyme replacement therapy targeting both lysosomal and cytoplasmic glycogen in Pompe disease.J Mol Med (Berl). 2017 May;95(5):513-521. doi: 10.1007/s00109-017-1505-9. Epub 2017 Feb 2. J Mol Med (Berl). 2017. PMID: 28154884
-
Adjunctive β2-agonists reverse neuromuscular involvement in murine Pompe disease.FASEB J. 2013 Jan;27(1):34-44. doi: 10.1096/fj.12-207472. Epub 2012 Sep 19. FASEB J. 2013. PMID: 22993195 Free PMC article.
References
-
- Hirschhorn R, Reuser AJJ. Glycogen Storage Disease Type II: Acid α-Glucosidase (Acid Maltase) Deficiency. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Basis for Inherited Disease. 8th ed. McGraw-Hill; New York: 2001. pp. 3389–419.
-
- Kishnani PS, Hwu WL, Mandel H, Nicolino M, Yong F, Corzo D. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr. 2006;148:671–6. - PubMed
-
- Engel AG, Gomez MR, Seybold ME, Lambert EH. The spectrum and diagnosis of acid maltase deficiency. Neurology. 1973;23:95–106. - PubMed
-
- Martiniuk F, Mehler M, Tzall S, Meredith G, Hirschhorn R. Sequence of the cDNA and 5'-flanking region for human acid alpha-glucosidase, detection of an intron in the 5' untranslated leader sequence, definition of 18-bp polymorphisms, and differences with previous cDNA and amino acid sequences. DNA Cell Biol. 1990;9:85–94. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
