

Cellular and molecular consequences of defective Fanconi anemia proteins in replication-coupled DNA repair: mechanistic insights
- PMID: 19622404
- PMCID: PMC2714807
- DOI: 10.1016/j.mrfmmm.2009.02.003
Cellular and molecular consequences of defective Fanconi anemia proteins in replication-coupled DNA repair: mechanistic insights
Abstract
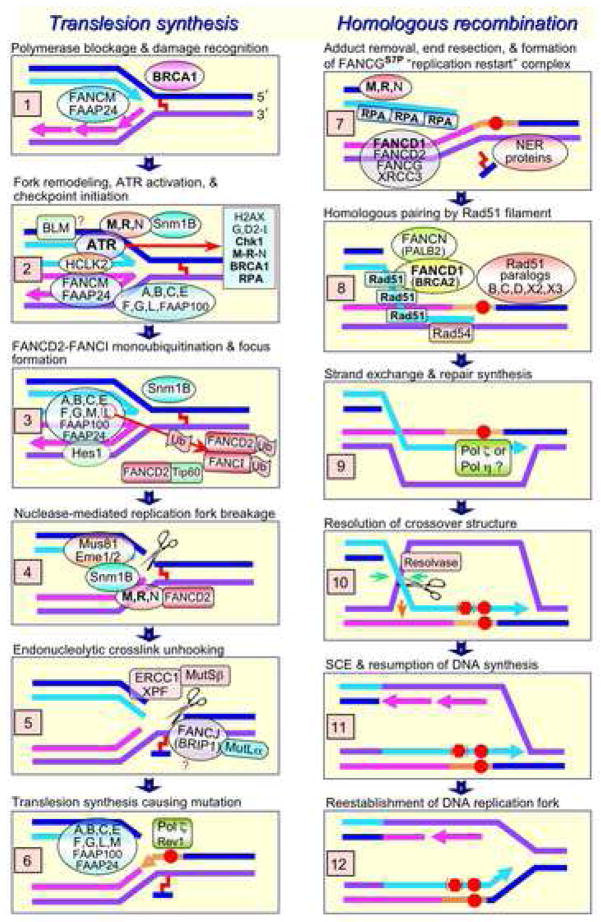
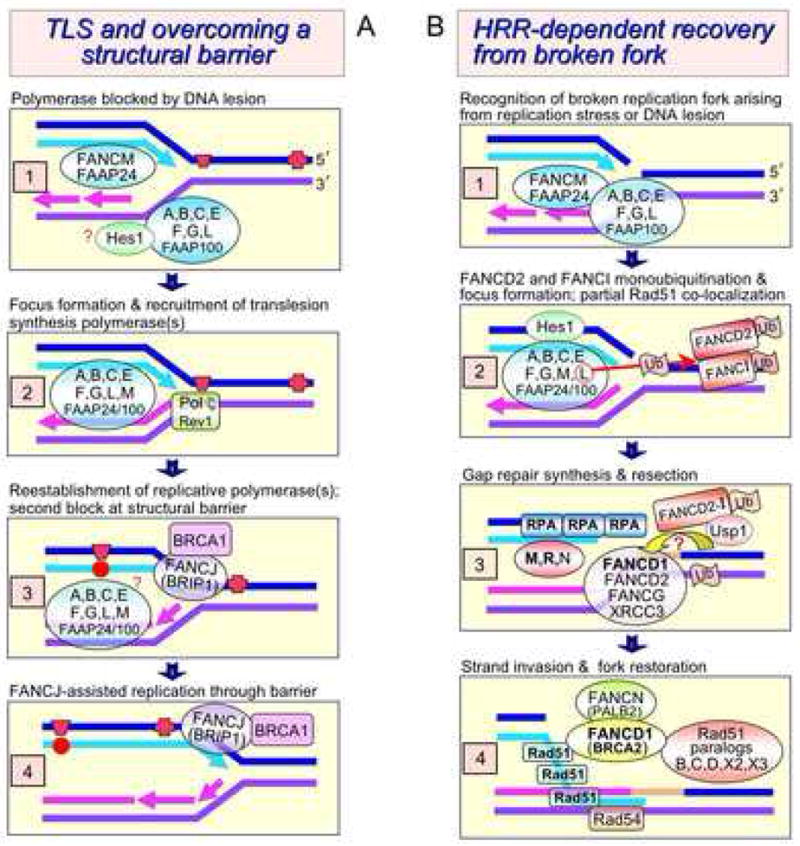
The Fanconi anemia (FA) molecular network consists of 15 "FANC" proteins, of which 13 are associated with mutations in patients with this cancer-prone chromosome instability disorder. Whereas historically the common phenotype associated with FA mutations is marked sensitivity to DNA interstrand crosslinking agents, the literature supports a more global role for FANC proteins in coping with diverse stresses encountered by replicative polymerases. We have attempted to reconcile and integrate numerous observations into a model in which FANC proteins coordinate the following physiological events during DNA crosslink repair: (a) activating a FANCM-ATR-dependent S-phase checkpoint, (b) mediating enzymatic replication-fork breakage and crosslink unhooking, (c) filling the resulting gap by translesion synthesis (TLS) by error-prone polymerase(s), and (d) restoring the resulting one-ended double-strand break by homologous recombination repair (HRR). The FANC core subcomplex (FANCA, B, C, E, F, G, L, FAAP100) promotes TLS for both crosslink and non-crosslink damage such as spontaneous oxidative base damage, UV-C photoproducts, and alkylated bases. TLS likely helps prevent stalled replication forks from breaking, thereby maintaining chromosome continuity. Diverse DNA damages and replication inhibitors result in monoubiquitination of the FANCD2-FANCI complex by the FANCL ubiquitin ligase activity of the core subcomplex upon its recruitment to chromatin by the FANCM-FAAP24 heterodimeric translocase. We speculate that this translocase activity acts as the primary damage sensor and helps remodel blocked replication forks to facilitate checkpoint activation and repair. Monoubiquitination of FANCD2-FANCI is needed for promoting HRR, in which the FANCD1/BRCA2 and FANCN/PALB2 proteins act at an early step. We conclude that the core subcomplex is required for both TLS and HRR occurring separately for non-crosslink damages and for both events during crosslink repair. The FANCJ/BRIP1/BACH1 helicase functions in association with BRCA1 and may remove structural barriers to replication, such as guanine quadruplex structures, and/or assist in crosslink unhooking.
Conflict of interest statement
Conflict of Interest
None
Figures
References
-
- Fanconi G. [Hypothesis of chromosomal translocation as a genetic interpretation of Fanconi’s familial constitutional panmyelopathy] Helv Paediatr Acta. 1964;19:29–33. - PubMed
-
- Alter BP. Cancer in Fanconi anemia, 1927–2001. Cancer. 2003;97:425–440. - PubMed
-
- Mathew CG. Fanconi anaemia genes and susceptibility to cancer. Oncogene. 2006;25:5875–5884. - PubMed
-
- Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat Rev Genet. 2007;8:735–748. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
