

Atypical Rett syndrome with selective FOXG1 deletion detected by comparative genomic hybridization: case report and review of literature
- PMID: 19623215
- PMCID: PMC2987032
- DOI: 10.1038/ejhg.2009.95
Atypical Rett syndrome with selective FOXG1 deletion detected by comparative genomic hybridization: case report and review of literature
Abstract
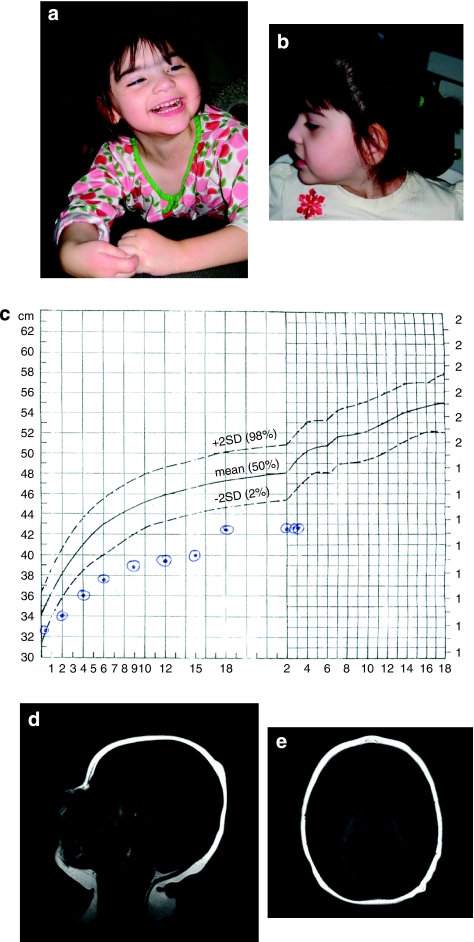
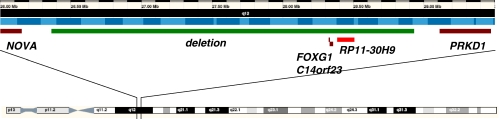
Rett syndrome is a severe neurodegenerative disorder characterized by acquired microcephaly, communication dysfunction, psychomotor regression, seizures and stereotypical hand movements. Mutations in methyl CpG binding protein 2 (MECP2) are identified in most patients with classic Rett syndrome. Genetic studies in patients with a Rett variant have expanded the spectrum of underlying genetic etiologies. Recently, a deletion encompassing several genes in the long arm of chromosome 14 has been associated with the congenital Rett-syndrome phenotype. Using array-based comparative genomic hybridization, we identified a 3-year-old female with a Rett-like syndrome carrying a de novo single-gene deletion of FOXG1. Her presentation included intellectual disability, epilepsy and a Rett-like phenotype. The variant features included microcephaly at birth and prominent synophrys. Our results confirm that congenital Rett syndrome can be caused by copy-number variation in FOXG1 and expand the clinical phenotypic spectrum of FOXG1 defect in humans.
Figures
References
-
- Rett A. [On a unusual brain atrophy syndrome in hyperammonemia in childhood] 1966;166:723–726. - PubMed
-
- Hagberg B. Rett syndrome: clinical peculiarities and biological mysteries. Acta Paediatr. 1995;84:971–976. - PubMed
-
- Hagberg B. Clinical delineation of Rett syndrome variants. Neuropediatrics. 1995;26:62. - PubMed
-
- Hagberg BA, Skjeldal OH. Rett variants: a suggested model for inclusion criteria. Pediatr Neuroly. 1994;11:5–11. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
